Hintergrund
Die Hashimoto-Enzephalopathie (HE) ist ein seltenes neuropsychiatrisches Syndrom, das mit Schilddrüsenantikörpern assoziiert ist und erstmals 1996 von Brain berichtet wurde (Brain et al., 1966). ER weist eine breite Palette klinischer Symptome auf, darunter neurologische Manifestationen wie schlaganfallähnliche Episoden, Krampfanfälle, Verwirrtheit, Myoklonus, Ataxie, Zittern und Demenz sowie psychiatrische Manifestationen wie akute Psychosen, depressive Störungen, Persönlichkeitsveränderungen, Halluzinationen und Schizophrenie (Kirshner, 2014; Menon et al., 2017). In diesem Fallbericht beschreiben wir einen Patienten mit HE, dessen klinische Symptome und Labortestergebnisse eine virale Enzephalitis nachahmten.
Fallbericht
Ein 59-jähriger Mann, der Fieber, Kopfschmerzen und unangenehme Sprache hatte, die sich speziell als langsame und unklare Sprache manifestierte, wurde ins Krankenhaus eingeliefert. Er bestritt die jüngsten Infektionen wie Grippe oder Gastroenteritis, Reisen und andere mögliche Gründe, die für das Fieber verantwortlich sein könnten, das bei 39,5 ° C seinen Höhepunkt erreichte. Seine neurologische Untersuchung und Schädel-Computertomographie (CT) und Magnetresonanztomographie (MRT) (Abbildung 1) Scans waren normal. Die EEG-Ergebnisse zeigten geringfügige Unregelmäßigkeiten in den Wellen (5-20 µv 14-20 Hz β), die von den bilateralen Hemisphären emittiert wurden. Blutroutine, C-reaktives Protein, Serum-Vitamin B12 und Folsäure sowie andere Autoimmunitätshersteller, die antinukleäre Antikörper (ANA), anti-neutrophile zytoplasmatische Antikörper (ANCA) und Rheumafaktoren enthielten, waren alle unauffällig. Die Zerebrospinalflüssigkeit (CSF) zeigte eine erhöhte Anzahl weißer Blutkörperchen (WBC) (104 × 106 / l, Referenzbereich 0-8 × 10 ^ 6 / l) und erhöhte Proteinspiegel (1,68 g / l, Referenzbereich 0,15–0,45 g / l). Kultur-, Abstrich- und Bakterien-, Pilz-, Virus- und Tuberkelbazillus-Antikörper im Serum und Liquor waren negativ. Bei ihm wurde eine virale Enzephalitis diagnostiziert und mit antiviralen Mitteln behandelt. Seine Symptome ließen innerhalb einer Woche nach und er wurde aus dem Krankenhaus entlassen. Fünf Monate später wurde er wegen eines Fiebers von 38,5 ° C und gelegentlicher Satzverwirrung erneut in unser Krankenhaus überwiesen. Eine weitere Lumbalpunktion wurde durchgeführt; Der Liquor hatte eine Leukozytenzahl von 39 × 10 ^ 6 / l und der Proteingehalt betrug 1,32 g / l. Die Ergebnisse der kranialen MRT und der Labortests waren fast normal, mit Ausnahme einer verminderten Schilddrüsenfunktion und erhöhter Anti-Schilddrüsen-Autoantikörper (ATA) -Spiegel im Serum und LIQUOR. Die ersten Befunde waren wie folgt: Im Serum betrug die Konzentration des Schilddrüsen–stimulierenden Hormons (TSH) 43,39 uIU / ml (Referenzbereich (RR): 0,27–4,2 uIU / ml), die Konzentration des freien Triiodthyronins (FT3) betrug 2,89 pmol / ml (RR: 3,1–6,8 pmol / ml), die Konzentration des freien Thyroxins (FT4) betrug 7,18 pmol / ml (RR: 12,0–22,0 pmol / ml), die die Trijodthyronin (TT3) -Konzentration betrug 1,17 pmol/ml (RR: 1,3-3.1 pmol/ml), and total thyroxine (TT4) concentration was 44.26 pmol/ml (RR: 66–181 pmol/ml); the titer of anti-thyroglobulin autoantibodies (TgAb) was 1274 IU/ml (RR: < 115 IU/ml) and the titer of anti-thyroperoxidase autoantibodies (TPOAb) was 600 IU/ml (RR: < 35 IU/ml). In the CSF, ATA was positive (TPOAb 17.06 IU/ml, TgAb 16.02 IU/ml). Ultrasound imaging indicated diffuse lesions of the thyroid. Antikörperanalysen von Anti-NMDAR, AMPA1, AMPA2, LGI1, CASPR2, GABA, GAD, Anti-Hu, Yo, Ri, MAI, MA2, CV2, Amphiphysin, SOX-1, Tr, Zic4 und GAD65 waren sowohl im Serum als auch im LIQUOR negativ. Bei dem Patienten wurde schließlich HE, Hashimoto-Thyreoiditis und Hypothyreose diagnostiziert und Methylprednisolon und Euthyrox verschrieben. Methylprednisolon wurde mit einer Dosis von 80 mg / Tag für 1 Woche begonnen und in der 2-Woche auf 40 mg / Tag reduziert; Anschließend wurde orales Prednisolon verschrieben, das mit einer Rate von 5 mg pro Woche entwöhnt wurde, dh orales Prednisolon wurde insgesamt 8 Wochen lang verwendet. Seine Symptome waren in 3 Tagen gelindert. Der Patient blieb während einer Nachbeobachtungszeit von 1 Jahr gesund.
ABBILDUNG 1
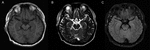
Abbildung 1. MRT-Bildgebung des Gehirns. Es gibt keine abnormalen Befunde in der MRT-Bildgebung des Gehirns des Patienten. (A) T1-gewichtetes Bild; (B) T2-gewichtetes Bild; (C) FLAIR-Bild.
Die geschätzte Prävalenzrate von HE beträgt 2,1 von 100.000 und das Geschlechterverhältnis (weiblich zu männlich) beträgt 4:1 (Ferracci et al., 2004). Der Verlauf der HE kann progressiv, rezidivierend oder sogar selbstlimitierend sein. Im vorliegenden Fall schien der Patient einen schubförmig-remittierenden Verlauf zu haben.
In Kombination mit den Symptomen des Patienten, normalen MRT-Ergebnissen, hohen ATA-Titern im Serum und ATA-Positivität des Liquors wurde bei dem Patienten schließlich HE diagnostiziert, nachdem andere mögliche Ursachen wie Schlaganfall, Tumor, Infektion des Zentralnervensystems, Autoimmunenzephalitis und paraneoplastisches Syndrom ausgeschlossen wurden.
Insgesamt ist die Pathophysiologie der HE noch nicht schlüssig. Aufgrund der neuropathologischen Befunde wurde ein autoimmuner Vaskulitismechanismus vorgeschlagen. In einer früheren Studie zeigten Gehirnbiopsien, dass eine lymphozytäre Infiltration um die Venolen und Arteriolen beteiligt sein kann (Duffey et al., 2003). Ein weiterer möglicher Mechanismus besteht darin, dass ATAs Antigene angreifen, die von der Schilddrüse und dem Gehirn gemeinsam genutzt werden, und aus diesem Grund hohe Titer von ATAs, einschließlich TPOAb-, TGAb- und Anti-TSH-Rezeptor (TSH-R) -Antikörpern im Serum und manchmal im LIQUOR, gelten als Markenzeichen von HE (Yoneda, 2018). In ähnlicher Weise wurden in unserem Fall hohe Titer von ATA und CSF-ATA gefunden. In einer früheren Studie führte die Plasmaaustauschtherapie zu einer tiefgreifenden Verbesserung der Symptome der Patienten (Tran et al., 2018). Zusammengenommen legen diese Ergebnisse nahe, dass ATA eine wichtige Rolle bei HE spielt. Erhöhte ATA-Spiegel im Serum finden sich jedoch auch in der Allgemeinbevölkerung und sind besonders häufig bei älteren Menschen. Darüber hinaus hängt das Ausmaß der ATA-Erhöhung nicht mit dem Schweregrad der HE zusammen. Obwohl die ATA-Serumspiegel des Patienten nach der Methylprednisolontherapie immer noch hoch blieben, waren seine Symptome abgeklungen (Titer der Antithyreoglobulin-Autoantikörper (TGAb): 041 IE / ml, RR: < 115 IE/ml; Titer der Anti-Thyroperoxidase-Autoantikörper (TPOA): 600 IE/ml, RR: < 35 IE/ml). Unsere Ergebnisse bestätigen, dass es keinen Zusammenhang zwischen dem Ausmaß der ATA-Erhöhung und dem Schweregrad der HE gibt.
Obwohl die Korrelation zwischen ATA und HE noch unklar ist (Kirshner, 2014), wurden diagnostische Kriterien für HE 2016 von einem Expertenteam in Lancet Neurology vorgeschlagen. Die Kriterien sind wie folgt: (1) Enzephalopathie mit Anfällen, Myoklonus, Halluzinationen oder schlaganfallähnlichen Episoden; (2) subklinische oder leichte offene Schilddrüsenerkrankung (normalerweise Hypothyreose); (3) normale Befunde oder unspezifische Anomalien, die durch MRT des Gehirns gezeigt werden; (4) Vorhandensein von Schilddrüsenantikörpern im Serum (Schilddrüsenperoxidase, Thyreoglobulin); (5) Fehlen gut charakterisierter neuronaler Antikörper im Serum und LIQUOR; und (6) angemessener Ausschluss alternativer Ursachen. ER kann diagnostiziert werden, wenn ein Patient alle Kriterien erfüllt (Graus et al., 2016). Unser Patient erfüllte alle oben genannten Kriterien und konnte daher mit HE diagnostiziert werden. Die meisten Patienten mit HE sprechen gut auf Steroide an; wenn Patienten jedoch steroidresistent sind, haben sich andere Immunsuppressionstherapien wie Plasmaaustausch, IVIg, Methotrexat und Mycophenolat als wirksam erwiesen. Hohe Dosen von Methylprednisolon (500-1000 mg) werden am häufigsten verwendet. Da der Patient leichte Symptome zeigte und die Verwendung einer hohen Dosis Methylprednisolon ablehnte, verordneten wir eine niedrige Dosis Methylprednisolon. Unser Patient unterzog sich einer Steroidtherapie und ist seitdem gesund geblieben. Der Patient erhielt während der Nachsorge keine anderen Immuntherapiebehandlungen. In einer retrospektiven Beobachtungsstudie von Mamoudjy et al. (2013) die Forschungsergebnisse zeigten, dass einige HE-Patienten einen Rückfall erlitten, sogar einen dritten Anfall, der eine kürzere Intervallzeit (durchschnittlich 18 Tage) im Vergleich zu der des zweiten Anfalls (durchschnittlich 213 Tage) aufwies. Patienten mit Rückfällen benötigten erneut eine Methylprednisolon- oder immunsuppressive Therapie (Mamoudjy et al., 2013). Unser Patient erlitt etwa 150 Tage nach dem ersten Anfall einen Rückfall und die Steroidtherapie war wirksam. Folgen wie Kopfschmerzen, Gedächtnisstörungen usw. waren ebenfalls häufig (Mamoudjy et al., 2013) hatte unser Patient jedoch glücklicherweise keine Folgen mehr.
Die Hashimoto-Enzephalopathie ist aufgrund ihrer Assoziation mit einem breiten Spektrum klinischer Symptome sowie ihrer unspezifischen Bildgebung und Elektroenzephalogrammdarstellung schwer genau zu diagnostizieren. Ein früherer Artikel berichtete über einen Fall von HE, der ursprünglich als virale Enzephalitis fehldiagnostiziert wurde (He et al., 2013), bei dem der Patient eine fortschreitende Beeinträchtigung der kognitiven Funktion und unkontrollierte Anfälle ohne Fieber aufwies. In unserem Fall führen die erhöhte Leukozytenzahl und das Liquorprotein in Kombination mit Fieber und Kopfschmerzen dazu, dass wir sie als virale Enzephalitis diagnostizieren. Fieber wurde bei mehreren Patienten mit HE mit und ohne Schilddrüsenerkrankungen beschrieben (Huang et al., 2011; Lu et al., 2015). Obwohl der Grund für Fieber in HE unklar ist, Lu et al. (2015) schlugen vor, dass es eine direkte Folge einer Entzündung in Kombination mit der Autoimmunvaskulitis sein könnte.
Die Hashimoto-Enzephalopathie kann sich auf viele verschiedene Arten manifestieren, darunter schlaganfallähnliche Episoden, Krampfanfälle, Verwirrtheit, Myoklonus, akute Psychosen, depressive Störungen und Halluzinationen, wodurch ER mit anderen Krankheiten verwechselt wird. Uwatoko et al. (2018) berichteten über einen Fall eines Patienten mit Parkinsonismus und einer tumorähnlichen Läsion, die durch MRT des Gehirns aufgedeckt wurde. Nach der Biopsie wurde festgestellt, dass die Läsion kein Tumor war, und ER wurde schließlich bestätigt (Uwatoko et al., 2018). Patienten mit HE können auch ungewöhnliche Symptome wie Pseudobulbärparese, sensomotorische Polyneuropathie, katatonische Symptome, Schwindel, Muskelschwäche, Chorea, Opsoclonus und Kopfschmerzen vom Typ Trigeminusneuralgie aufweisen (Beckmann et al., 2011; Salazar et al., 2012; Scharan et al., 2015; Ueno et al., 2016; Karthik et al., 2017; Emeksiz et al., 2018; Oz Tuncer et al., 2018). Schnell fortschreitende Demenz als häufige Manifestation von HE macht es notwendig, HE von anderen Krankheiten zu unterscheiden, die durch vaskuläre, infektiöse, toxisch-metabolische und autoimmune Faktoren, Metastasen / Neoplasien, iatrogene / angeborene Stoffwechselfehler, neurodegenerative Erkrankungen und systemische Erkrankungen / Anfälle verursacht werden (Paterson et al., 2012).
Schlussfolgerung
Zusammenfassend ist die Diagnose von HE ziemlich komplex, aber wertvoll wegen seiner dramatischen Reaktion auf immunsuppressive Therapie. Wenn Kliniker mit ungeklärter Enzephalitis konfrontiert sind, sollten Schilddrüsenfunktion und ATA-Spiegel als konventionelle Tests betrachtet werden. ER kann als Diagnose für Patienten mit normalen ATA-Serumspiegeln ausgeschlossen werden. Bei Patienten mit erhöhten ATA-Spiegeln sollte ER in Betracht gezogen werden, nachdem andere mögliche Krankheiten ausgeschlossen wurden.
Data Availability Statement
Die für diese Studie generierten Datensätze stehen dem korrespondierenden Autor auf Anfrage zur Verfügung.
Ethikerklärung
Die Studien mit menschlichen Teilnehmern wurden von der Ethikkommission des Ersten Krankenhauses der Universität Jilin, China, überprüft und genehmigt. Die Patienten / Teilnehmer gaben ihre schriftliche Einverständniserklärung zur Teilnahme an dieser Studie ab. Für die Veröffentlichung potenziell identifizierbarer Bilder oder Daten, die in diesem Artikel enthalten sind, wurde von der / den Person (en) eine schriftliche Einverständniserklärung eingeholt.
Autorenbeiträge
HM hat an der Konzeption und Gestaltung des Manuskripts mitgewirkt. Ich schrieb den ersten Entwurf des Manuskripts. YY, XM, YX und NS schrieben Abschnitte des Manuskripts. Alle Autoren trugen zur Überarbeitung des Manuskripts bei, lesen, und genehmigte die eingereichte Version.
Finanzierung
Diese Arbeit wurde vom National Key R&D Program of China (Nr. 2017YFC0110304) unterstützt.
Interessenkonflikt
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Beckmann, Y. Y., Top, D. und Yigit, T. (2011). Ungewöhnliche Präsentationen der Hashimoto-Enzephalopathie: Trigeminusneuralgieform Kopfschmerzen, Skew-Abweichung, Hypomanie. Seite 40, 495-496. ust-idnr.: 10.1007/s12020-011-9506- x
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Brain, L., Jellinek, EH und Ball, K. (1966). Hashimoto-Krankheit und Enzephalopathie. Lanzette 2, 512-514.
Google Scholar
Duffey, P., Yee, S., Reid, I. N. und Bridges, L. R. (2003). Hashimoto-Enzephalopathie: postmortale Befunde nach tödlichem Status epilepticus. Neurologie 61, 1124-1126.
PubMed Zusammenfassung / Google Scholar
Emeksiz, S., Kutlu, N. O., Alacakir, N. und Caksen, H. (2018). Ein Fall von steroidresistenter Hashimoto-Enzephalopathie mit sensomotorischer Polyneuropathie. Türke. In: J. Pediatr. 60, 310–314. Ursprungsbezeichnung: 10.24953/turkjped.2018.03.012
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Ferracci, F., Bertiato, G. und Moretto, G. (2004). Hashimoto-Enzephalopathie: epidemiologische Daten und pathogenetische Überlegungen. J. Neurol. Sci. 217, 165–168.
PubMed Zusammenfassung / Google Scholar
Graus, F., Titulaer, M. J., Balu, R., Benseler, S., Bien, C. G., Cellucci, T., et al. (2016). Ein klinischer Ansatz zur Diagnose von Autoimmun-Enzephalitis. In: Lancet Neurol. 15, 391–404.
Google Scholar
Er, L., Li, M., Lang, XH, Li, XPUND Peng, Y. (2013). Ein Fall von Hashimoto-Enzephalopathie, der fälschlicherweise als virale Enzephalitis diagnostiziert wurde. Uhr. J. Rechtssache Rep. 14, 366-369.
Google Scholar
Huang, W., Xia, C. und Chatham, M. (2011). Infektionskrankheit oder Hashimoto-Enzephalopathie fackeln: ein Fallbericht. Seite 20, 717-719. doi: 10.1016/j.Beschlagnahme.2011.04.011
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Karthik, MS, Nandhini, K., Subashini, V. und Balakrishnan, R. (2017). Hashimoto-Enzephalopathie mit ungewöhnlichen Verhaltensstörungen bei einem jugendlichen Mädchen. Rechtssache Rep. Med. 2017, 3494310. doi: 10.1155/2017/3494310
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Kirshner, H. S. (2014). Hashimoto-Enzephalopathie: ein kurzer Rückblick. Curr. Neurol. Neurowissenschaften. 14:476.
Google Scholar
Lu, T., Zhou, Z., Wu, A., Qin, B. und Lu, Z. (2015). Febrile Hashimoto-Enzephalopathie im Zusammenhang mit Hashitoxikose. In: Acta Neurol. Belg. 115, 811–813.
Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitationen Mamoudjy, N., Korff, C., Blanchard, G., Loiseau-Corvez, M. N., et al. (2013). Hashimoto-Enzephalopathie: Identifizierung und langfristiges Ergebnis bei Kindern. EUR. In: J. Paediatr. Neurol. 17, 280–287. Ursprungsbezeichnung: 10.1016/j.ejpn.2012.11.003
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Menon, V., Subramanian, K. und Thamizh, JS (2017). Psychiatrische Präsentationen, die die Hashimoto-Enzephalopathie ankündigen: eine systematische Überprüfung und Analyse der in der Literatur berichteten Fälle. In: J. Neurosci. Ländlich. Prakt. 8, 261–267. doi: 10.4103/jnrp.jnrp_440_16
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Oz Tuncer, G., Teber, S., Kutluk, M. G., Albayrak, P. und Deda, G. (2018). Hashimoto-Enzephalopathie als Pseudobulbärparese. Kinder Nerv. Syst. 34, 1251–1254. ust-idnr.: 10.1007/s00381-018-3720-2
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Paterson, R. W., Takada, L. T. und Geschwind, M. D. (2012). Diagnose und Behandlung von schnell fortschreitenden Demenzen. In: Neurol Clin. Prakt. 2, 187–200.
PubMed Zusammenfassung / Google Scholar
Salazar, R., Mehta, C., Zaher, N. und Miller, D. (2012). Opsoclonus als Manifestation der Hashimoto-Enzephalopathie. J. Clin. Neurowissenschaften. 19, 1465–1466. doi: 10.1016/j.jocn.2012.02.012
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Sharan, A., Sengupta, S., Mukhopadhyay, S. und Ghosh, B. (2015). Hashimoto-Enzephalopathie mit Chorea. J. Assoc. Ärzte Indien 63, 83-84.
PubMed Zusammenfassung / Google Scholar
Tran, M. H., Mkhikian, H., Sy, M., Perez-Alvarez, I. und Demetriou, M. (2018). Langfristiger Plasmaaustausch als Erhaltungstherapie bei Hashimoto-Enzephalopathie vom Kleinhirntyp, ein Fallbericht. Transfusion. Apher. Sci. 57, 418–420. doi: 10.1016/j.Abschrift.2018.05.027
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Ueno, H., Nishizato, C., Shimazu, T., Watanabe, H., Mizukami, T., Kosuge, H., et al. (2016). Hashimoto-Enzephalopathie mit Schwindel und Muskelschwäche bei einem männlichen pädiatrischen Patienten. Keine Hattatsu 48, 45-47.
PubMed Zusammenfassung / Google Scholar
Uwatoko, H., Yabe, I., Sato, S., Abe, M., Shirai, S., Takahashi, I., et al. (2018). Hashimoto-Enzephalopathie imitiert einen Gehirntumor und seine pathologischen Befunde: ein Fallbericht. J. Neurol. Sci. 394, 141–143. Ursprungsbezeichnung: 10.1016/j.jns.2018.09.008
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Yoneda, M. (2018). Hashimoto-Enzephalopathie und Autoantikörper. Gehirnnerv 70, 305-314. Ursprungsbezeichnung: 10.11477/mf.1416201004
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar