Im März 2017 widmete sich ein Mini-Symposium auf dem 11. Kongress für Kontroversen in der Neurologie (CONy), Athen, Griechenland, der Neuro-Behçet-Krankheit (NBD). Einer Einführung in die wichtigsten klinischen Merkmale der Behçet-Krankheit (BD) folgten eine Überprüfung ihrer neurologischen Manifestationen und eine fokussierte Darstellung der Herausforderungen der Differentialdiagnose. Dieser Rückblick stellt einen Bericht des Mini-Symposiums in Form einer aktualisierten Zusammenfassung der vorgestellten Themen dar. Es folgt der Struktur des Mini-Symposiums und erweitert teilweise die dort präsentierten Informationen. Eine systematische Überprüfung geht jedoch über den Rahmen dieses Berichts hinaus. Die allgemeine Einführung in BD wird kurz und bündig sein, da der Schwerpunkt dieser Überprüfung auf NBD liegt. Details zu BD im Allgemeinen finden Sie in aktuellen Reviews, zum Beispiel Yazici et al.1
Morbus Behçet
Geschichte und Epidemiologie
BD ist eine chronische, multisystemische und polysymptomatische Erkrankung mit unvorhersehbaren Exazerbationen und Remissionen. Alle Systeme können gleichzeitig oder nacheinander betroffen sein.1 Es gibt mehrere klinische Untergruppen und geografische Unterschiede, die auf unterschiedliche Krankheitsmechanismen hinweisen. In Anbetracht dessen bevorzugen einige Autoren die Verwendung des Begriffs Behçet-Syndrom anstelle der Behçet-Krankheit. Aus Gründen der Kohärenz und Harmonisierung und um der Terminologie der Diskussionen des Mini-Symposiums zu folgen, wird in dieser Übersicht der Begriff Morbus Behçet verwendet. Im Jahr 1930 beschrieb der Augenarzt Adamantiades die Koexistenz von Augen-, Haut- und Schleimhautläsionen bei einem Patienten.2 1937 identifizierte Hulusi Behçet BD bei zwei Patienten, die zusätzlich zu einer Augenerkrankung an Mund- und Genitalgeschwüren litten, und beschrieb sie als separate Krankheit, von der angenommen wurde, dass sie durch ein Virus verursacht wird.3
Epidemiologische Studien haben eine große Variabilität in der Prävalenz von BD in Abhängigkeit von der geografischen und ethnischen Herkunft der Bevölkerung gezeigt. Es gibt Hinweise darauf, dass die Prävalenz von BD in den Gebieten höher ist, in denen die Bevölkerung eine hohe Inzidenz des HLA-B51-Allels aufweist.4 Es wird oft als Seidenstraßenkrankheit bezeichnet, da es in Ländern des Mittelmeers, des Nahen Ostens und des Fernen Ostens auf der alten Seidenhandelsroute relativ häufig vorkommt. Die Türkei hat mit 119,8 pro 100.000 die höchste Prävalenz.4
Pathophysiologie
Die Pathomechanismen von BD sind nicht vollständig bekannt; Es kann jedoch als ein Zustand angesehen werden, der mit Autoimmunreaktionen, Autoinflammation und Gefäßverletzungen verbunden ist. Die Anfälligkeit scheint durch ein Zusammenspiel von genetischen und Umweltfaktoren bestimmt zu werden. Unter den Suszeptibilitätsgenen wurden solche für Zytokine wie Interleukin (IL) -17, IL-12, IL-23, IL-21, IL-23, Tumornekrosefaktor (TNF) -α, IL-1β und IL-8 in Verbindung gebracht.5 Eine Fehlregulation dieser entzündungsfördernden Zytokine kann eine unkontrollierte Aktivierung des angeborenen Immunsystems mit oder ohne Aktivierung der adaptiven Immunantworten widerspiegeln, die für die pathologischen Merkmale verantwortlich zu sein scheinen. Eine erhöhte Produktion von entzündungsfördernden Zytokinen wie TNF-α, IL-1β und IL-8 führt zur Aktivierung von Neutrophilen und zur Verstärkung der zellulären Wechselwirkungen zwischen Neutrophilen und Endothelzellen.6 Diese aktivierten Neutrophilen produzieren übermäßige Superoxide und lysosomale Enzyme, die zu Gewebeverletzungen führen. Die resultierenden Läsionen sind histologisch durch neutrophile angiozentrische Infiltrate mit leukozytoklastischer (früher) oder lymphozytärer (später) Vaskulitis mit oder ohne murale Thrombose und Nekrose gekennzeichnet.7 Darüber hinaus wurde berichtet, dass die B-Lymphozytenfunktion bei einigen Patienten abnormal ist.8
Zu den Umweltfaktoren, die für die Anfälligkeit für BD verantwortlich sind, gehören am häufigsten Bakterien wie Streptococcus sanguinisund Viren, hauptsächlich Herpesviren. Tatsächlich wird die Gen–Umwelt-Interaktion bei BD durch dichte Genotypisierungsstudien angezeigt, die eine dysregulierte Wirtsimmunantwort auf bakterielle Antigene mit der BD-Anfälligkeit in Verbindung bringen.9
Klinische Manifestationen und Diagnose
BD hat eine breite Palette von klinischen Manifestationen. Mundgeschwüre sind normalerweise das erste Symptom; Sie können Jahre vor der Diagnose auftreten und sind während des Krankheitsverlaufs mit einer Häufigkeit von fast 100% vorhanden. Bei 75% der Patienten treten anogenitale Aphten auf, hauptsächlich am Hodensack und Penis bei Männern und an der Vulva bei Frauen. Bei etwa 60% der Patienten wurden verschiedene Hautläsionen berichtet, darunter Erythema nodosum, papullopustulöse Läsionen, Pseudofollikulitis, Pyoderma gangrenosum und kutane Vaskulitis.1,10
Darüber hinaus sind zahlreiche andere Bereiche häufig von BD betroffen. Okuläre Darstellungen von BD treten bei 30-80% der Patienten (vorwiegend bei Männern) auf und sind eine Hauptursache für Morbidität, da sie insbesondere bei Netzhautvaskulitis zur Erblindung führen können.1 Gemeinsame Beteiligung wird auch häufig in BD berichtet. Mono / Polyarthritis bei Kindern ist Kursivnicht erosiv und die am stärksten betroffenen Gelenke sind Knie, Knöchel, Füße und Kursivhände. Die gastrointestinale Beteiligung ist durch Schmerzen, Blutungen, Darmschleimhautulzerationen oder Darmperforationen gekennzeichnet. ItalicsVascular Beteiligung kann auch auftreten, am häufigsten tiefe / oberflächliche periphere Venenthrombose und auch aneurysmatische / okklusive arterielle Verschlusskrankheit. Die Herzbeteiligung umfasst Koronararteriitis, Gefäßerkrankungen, intrakardiale Thromben oder Thrombosen venöser Kollateralgefäße und Vena cava superior, Myokarditis und rezidivierende ventrikuläre Arrhythmien.1 Neurologische Beteiligung ist ebenfalls eine häufige Manifestation von BD und wird im folgenden Abschnitt weiter erörtert.
Die diagnostischen Kriterien für BD wurden vor 28 Jahren veröffentlicht.11 Das Hauptkriterium sind wiederkehrende orale Ulzerationen (aphthös oder herpetiform), die vom Arzt beobachtet oder vom Patienten mindestens dreimal in einem Zeitraum von 12 Monaten zuverlässig gemeldet werden. Zusätzlich müssen zwei der vier Nebenkriterien erfüllt sein:
- rezidivierende Genitalulzerationen;
- Augenläsionen: Uveitis anterior, Uveitis posterior, Zellen im Glaskörper durch Spaltlampenuntersuchung oder von einem Augenarzt beobachtete Netzhautvaskulitis;
- Hautläsionen: Erythema nodosum, Pseudofollikulitis, papulopustulöse Läsionen oder akneiforme Knötchen bei postjugendlichen Patienten, die nicht mit Kortikosteroiden behandelt wurden; und
- ein positiver pathologischer Test (Hautpricktest): unspezifische Hautkursivhyperreaktivität als Reaktion auf ein geringfügiges Trauma Lesen Sie ein Arzt nach 24-48 Stunden (> 2 mm Pustel, nach Unterarmhaut (5 mm Tiefe) mit 20-22 g Nadel stechen).
Behandlungsmöglichkeiten
Das Ziel der BD-Behandlung ist es, entzündliche Exazerbationen und Rezidive umgehend zu unterdrücken, um irreversible Organschäden zu verhindern, und ein multidisziplinärer Ansatz ist erforderlich. Die Behandlung von BD hängt davon ab, ob selbstlimitierende Manifestationen oder eine schwerwiegende Organbeteiligung vorliegen, und sollte nach Alter, Geschlecht und Art individualisiert werden.12,13 Bei systemischen schweren Erkrankungen können intravenöse Pulskortikosteroide, gefolgt von täglichen oralen Dosen, Azathioprin, Cyclophosphamid, Cyclosporin-A, Methotrexat, Mycophenolatmofetil, Tacrolimus, Interferon (IFN) -α- oder TNF-α-Inhibitoren (Etanercept, Infliximab) gewählt werden.12 Ein neuer oraler Inhibitor von Phosphodiesterase-4, Apremilast, wurde bei Patienten ohne größere Organbeteiligung untersucht und zeigte bei signifikant mehr Patienten ein vollständiges Ansprechen.14
Andere Behandlungen zielen darauf ab, die spezifischen Symptome von BD zu kontrollieren. TNF-α-Inhibitoren sind bei BD hochwirksam, insbesondere bei Augenbeteiligung.15,16,17 Die Langzeitanwendung des TNF-α-Inhibitors Infliximab kann die Häufigkeit von Augenrezidiven auch in resistenten Fällen verringern. Als Erstlinientherapie sollte Infliximab in Verbindung mit einem Immunsuppressivum (Azathioprin, Kortikosteroide oder Kursivmethotrexat) begonnen werden, und bei Auftreten einer Remission sollten gleichzeitig Kortikosteroide reduziert werden. Viele Studien haben die Wirksamkeit von Infliximab bei BD gezeigt.15-17 Bei Patienten mit Augenbeteiligung des hinteren Segments sollten Azathioprin, Cyclosporin-A, IFN-α oder Anti-TNF-α, häufig kombiniert mit systemischen Kortikosteroiden, eingeleitet werden. Bei Patienten mit posteriorer Augenbeteiligung sollten Azathioprin, Cyclosporin-A, IFN-α oder Anti-TNF-α, häufig kombiniert mit systemischen Kortikosteroiden, eingeleitet werden.12,13
Zur Behandlung schwerer Gefäßerkrankungen mit thrombotischen Ereignissen bei BD werden Kortikosteroide und Immunsuppressiva wie Azathioprin, Cyclophosphamid oder Cyclosporin-A empfohlen,12 und Kursivanti-TNF-α könnte bei refraktären Patienten in Betracht gezogen werden. Antikoagulanzien werden in Betracht gezogen, wenn das Blutungsrisiko im Allgemeinen gering ist und koexistierende Lungenarterienaneurysmen ausgeschlossen sind.12,13 Bei gastrointestinalen Symptomen können 5-Aminosalicylsäurederivate, einschließlich Sulfasalazin oder Mesalamin, systemische Kortikosteroide, Azathioprin, Anti-TNF-α und Thalidomid verwendet werden. Die Beteiligung des Zentralnervensystems, einschließlich akuter Anfälle einer zerebralen parenchymalen Beteiligung, wird mit hochdosierten Kortikosteroiden behandelt, gefolgt von einer Verjüngung zusammen mit Immunsuppressiva. Obwohl Cyclosporin kostengünstig ist, sollte es aufgrund des Risikos einer Neurotoxizität vermieden werden. Anti-TNF-α sollte bei schwerer oder refraktärer Erkrankung als Italicsfirst-line in Betracht gezogen werden. Die erste Episode einer zerebralen Venenthrombose (CVT) sollte mit hochdosierten Kortikosteroiden behandelt werden, gefolgt von einer Verjüngung. Antikoagulanzien können für kurze Zeit hinzugefügt werden.
Neurologische Beteiligung an Morbus Behçet
Wie bei systemischen Erkrankungen sind die neurologischen Manifestationen von BD vielfältig.18-22 Neurologische Beteiligung an BD kann klassifiziert werden als; 1) primär, bei dem die neurologische Beteiligung direkt auf BD zurückzuführen ist und als NBD oder Neuro-Behçet-Syndrom (NBS) bezeichnet wird; und 2) sekundär, bei dem die neurologischen Manifestationen das Ergebnis neurologischer Komplikationen sind, die auf eine systemische Beteiligung von BD zurückzuführen sind (d. H. zerebrale Embolien aufgrund kardialer Komplikationen der BD, erhöhter Hirndruck infolge des Syndroms der oberen Hohlvene) oder aufgrund der Therapien, die für die systemischen Manifestationen der BD angewendet werden (d. H. Neurotoxizität des Zentralnervensystems durch Cyclosporin; periphere Neuropathie infolge von Thalidomid oder Colchicin). Dies sind indirekte Ursachen für neurologische Probleme bei Patienten mit BD und werden nicht als NBD bezeichnet.
Eine primäre neurologische Beteiligung an NBD tritt bei bis zu 10% aller Patienten auf.18-22 NBD tritt normalerweise innerhalb des vierten Jahrzehnts und ungefähr 5 Jahre nach Beginn der systemischen Erkrankung auf. Obwohl einige Patienten eine neurologische Beteiligung aufweisen können, ohne die Klassifizierungskriterien der International Study Group (ISG) für BD zu erfüllen11 und eine Diagnose von NBD kann typischerweise nicht gestellt werden, es sei denn, es liegen mindestens die Vorgeschichte oder die Folgen einiger systemischer Manifestationen von BD vor. Obwohl BD bei beiden Geschlechtern fast gleich häufig auftritt, ist NBD bei Männern häufiger (3: 1).23 BD und NBD sind in der pädiatrischen Population selten; wenn jedoch neurologische Beteiligung bei Kindern auftritt, ist es oft bald nach dem Beginn der systemischen Erkrankung.20.21
Klinische und bildgebende Befunde weisen darauf hin, dass es zwei Hauptformen von NBD gibt: 1) entzündliche parenchymale Erkrankung des ZNS (p-NBD); und seltener 2) eine extraparenchymale Form (ep-NBD), an der große extraparenchymale Gefäßstrukturen, hauptsächlich venöse Duralhöhlen, beteiligt sind, die eine zerebrale venöse Sinusthrombose (CVST) verursachen. Im Gegensatz zu p-NBD wird CVST häufiger bei pädiatrischen Patienten mit NBD beobachtet, und diese beiden Arten der Beteiligung treten sehr selten bei derselben Person auf und weisen daher wahrscheinlich unterschiedliche Pathogenesen auf. In seltenen Fällen kann eine aseptische Meningitis die Präsentation von extraparenchymaler NBD sein. Verhaltens- und psychotische Symptome, die als ‚Neuro-Psycho-Behçet‘ bezeichnet werden, können im Verlauf der NBD beobachtet werden. Kognitive Funktionen sind wahrscheinlich auch in einer Untergruppe von Patienten mit BD betroffen, und frontale (exekutive) Dysfunktion ist das am häufigsten beobachtete Muster. Eine primäre Beteiligung des peripheren Nervensystems wurde bei BD berichtet, ist jedoch äußerst selten.18,19
Das häufigste neurologische Symptom bei NBD sind Kopfschmerzen, die sowohl bei parenchymaler (p-NBD) als auch bei extraparenchymaler NBD (ep-NBD, CVST) auftreten. Kopfschmerzen können jedoch auch ein Symptom einer schweren Augenentzündung sein; kann mit Exazerbationen der systemischen Symptome von BD verbunden sein, mit einigen migräneähnlichen Merkmalen, und wird als ’nichtstruktureller Kopfschmerz von BD‘ bezeichnet; oder kann unabhängig von BD sein und kann als primärer Kopfschmerz mit ähnlichen Raten wie die Allgemeinbevölkerung koexistieren. Andere häufige Symptome sind Schwäche (Hemiparese), Gangstörungen (Ataxie), Sprachschwierigkeiten (Dysarthrie) und seltener verhaltens- und kognitive Veränderungen. Sehverlust durch Optikusneuritis, sensorische und extrapyramidale Symptome und Krampfanfälle sind selten.18-22
Es wird vorgeschlagen, die Kriterien der International Consensus Recommendation (ICR) bei der Diagnose von NBD zu verwenden.24 Diese Kriterien können zusammengefasst werden als ‚das Auftreten neurologischer Symptome und Anzeichen bei einem Patienten, der die ISG-Diagnosekriterien für BD erfüllt, die nicht anderweitig durch eine andere bekannte systemische oder neurologische Erkrankung oder Behandlung erklärt werden und bei dem objektive Anomalien im Einklang mit NBD entweder bei neurologischen Untersuchungen, Neuroimaging-Studien, MRT oder abnormalen Untersuchungen der Liquor cerebrospinalis (CSF) festgestellt werden‘.24 Die ICR-Kriterien umfassen auch ein wahrscheinliches NBD-Kriterium, aber man sollte vorsichtiger sein, wenn man eine solche Diagnose stellt.
Parenchymale Neuro-Behçet-Krankheit
Etwa 75-80% der Patienten mit NBD weisen eine parenchymale Beteiligung auf. Die Hauptsymptome und Anzeichen von P-NBS sind Kopfschmerzen, Dysarthrie, Ataxie, Hemiparese und kraniale Neuropathien (hauptsächlich Beteiligung von motorischen Augen- und Gesichtsnerven), die sich normalerweise subakut entwickeln. p-NBD ist eine der Hauptursachen für Morbidität und Mortalität bei NBD. Ähnlich wie bei Multipler Sklerose (MS) kann der klinische Verlauf der p-NBD bei einem einzigen Anfall bestehen bleiben, eine rezidivierende Form haben oder progressiv sein. Patienten mit p-NBD haben jedoch in der Regel neurologische Defizite. Die Läsionen von p-NBD-Läsionen betreffen häufig den Telencephalic / Diencephalic-Übergang und den Hirnstamm, die normalerweise groß sind und keine deutlichen Grenzen aufweisen. In der akuten Phase verstärken sich diese Läsionen wahrscheinlich und treten im Allgemeinen in einem heterogenen Muster auf. Tumefactive zerebrale Läsionen können beobachtet werden, und Rückenmarksverletzungen, obwohl nicht häufig, wenn sie auftreten, sind wahrscheinlich in Längsrichtung ausgedehnt. Bei diesen Patienten fehlen typischerweise Anti-MOG- und AQP4-Antikörper. Eine fokale Beteiligung des Kleinhirns ist selten, es wurde jedoch über eine isolierte Kleinhirnatrophie berichtet.25 Läsionen in intraparenchymalen Hauptarterienarterien sind ungewöhnlich und die Beteiligung großer extraparenchymaler oder kleinerer intraparenchymaler Arterien, obwohl äußerst selten, wurde berichtet, was darauf hindeutet, dass eine Untergruppe der arteriellen p-NBD existieren kann.26 Aufgrund der radiologischen und histopathologischen Befunde ist eine venöse Pathogenese die wahrscheinliche Erklärung für p-NBS-Läsionen.17-19,26
Die Liquorbefunde können eine ausgeprägte Pleozytose und einen erhöhten Proteinspiegel während der akuten Episode von p-NBD offenbaren. Die neutrophile Vorherrschaft ist während der akuten Phase typisch, wird jedoch später durch eine lymphozytäre Form ersetzt. Oligoklonale Banden werden selten nachgewiesen.
Extraparenchymale Neuro-Behçet-Krankheit / zerebrale venöse Sinusthrombose
Bis zu 20% der Patienten mit NBD haben CVST. Diese Patienten haben starke Kopfschmerzen, die sich normalerweise über einige Wochen entwickeln. Typischerweise zeigt die funduskopische und neurologische Untersuchung ein Papillenödem und gelegentlich eine sechste Nervenlähmung. Im Vergleich zu anderen Ursachen für eine Duralsinusthrombose sind Bewusstseinsstörungen, fokale neurologische Defizite wie Hemiparese und epileptische Anfälle bei extraparenchymaler NBD selten und venöse Infarkte unwahrscheinlich. Eine Magnetresonanzvenographie bestätigt die Diagnose und zeigt das Ausmaß der CVST. Mit Ausnahme eines erhöhten Öffnungsdrucks sind die Liquorbefunde im Allgemeinen normal, außer in der seltenen meningitischen Präsentation, in der eine hohe Anzahl von Neutrophilen gefunden werden kann. Wie bereits erwähnt, tritt diese Form der NBD häufiger in der pädiatrischen Bevölkerung auf, was darauf hindeutet, dass das Alter die Form der neurologischen Beteiligung beeinflussen kann.
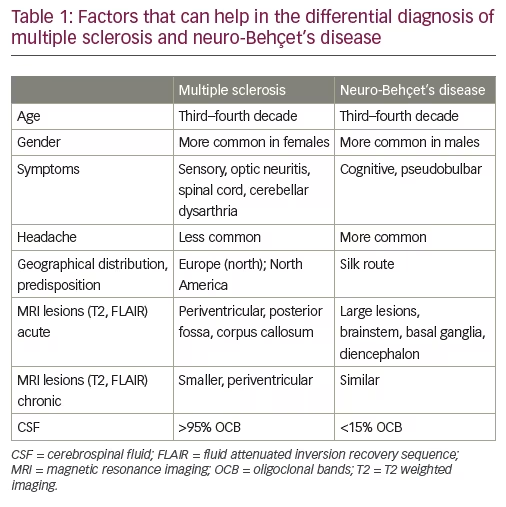
Neuro-Behçet-Krankheit – Differentialdiagnose
Die Differentialdiagnose von NBD bleibt eine große Herausforderung. Besonders schwierig sind Situationen, in denen ein Patient ohne ausgewachsene BD neurologische Manifestationen aufweist. Es ist wichtig zu wissen, dass neurologische Manifestationen von BD eine schwerere Erkrankung widerspiegeln und selten isoliert gesehen werden. Daher müssen Kliniker, die NBD vermuten, wachsam sein, um nach der wahrscheinlichen Entwicklung von Symptomen und Anzeichen außerhalb des ZNS zu suchen. Dies ist auch bei Patienten mit bekannter BD wichtig, da neurologische Manifestationen tendenziell bei schweren Schüben auftreten und systemische BD-Manifestationen gesucht werden sollten. Bei meningitischen Präsentationen ist die Differentialdiagnose einer akuten bakteriellen Meningitis unerlässlich. Akute bakterielle Meningitis ist ein medizinischer Notfall und Antibiotika-Behandlung wird oft in Notfallsituationen vor der mikrobiologischen Bestätigung eingeleitet. Obwohl der Liquor bei meningitischer NBD häufig weniger Leukozyten aufweist, kann er manchmal die Zahlen erreichen, die bei bakterieller Meningitis auftreten, und ein Versagen der Erkennung kann schwerwiegende Folgen haben. Eine vorübergehende klinische Verbesserung kann nach Einführung von unterstützenden Maßnahmen und Antibiotika beobachtet werden, die die aggressive Behandlung von BD verzögern können.
Uveomeningeale Syndrome sind eine heterogene Gruppe entzündlicher Erkrankungen, die durch meningeale Entzündung und Augenbeteiligung (Uveitis) gekennzeichnet sind.27 Die Differentialdiagnose umfasst viele Entitäten, und die damit verbundenen systemischen Merkmale, die spezifische Organbeteiligung (z. B. Lunge bei Sarkoidose) sowie Bildgebungs- und Biomarkermerkmale können dazu beitragen, NBD von den anderen Zuständen zu unterscheiden. Neben NBD umfassen uveomeningeale Syndrome:
- sarkoidose;
- Granulomatose mit Polyangiitis;
- Syphilis;
- Vogt-Koyanagi-Harada-Krankheit; und
- akute posteriore multifokale Placoidpigmentepitheliopathie.
Eine der wichtigsten und herausforderndsten Überlegungen bei der Differentialdiagnose von NBD ist MS.18,28 Wenn die Diagnose für beide Entitäten gut etabliert ist, ist die Unterscheidung relativ einfach; In den Anfangsphasen kann die Unterscheidung jedoch schwierig sein. Die parenchymalen Läsionen von NBD können in den meisten Aspekten die von MS im MRT nachahmen, einschließlich der eiförmigen Form der kallosalen Dawson-Finger und des Vorhandenseins einer zentralen Vene. Aufgrund der perivenulären Verteilung von Läsionen unter beiden Bedingungen, Der Nutzen des zentralen Zeichens und seine Rolle bei der Differentialdiagnose von MS versus NBD wird in Expertenkonsens weiterhin diskutiert.28,29 Während dies zu Beginn der Erkrankung besonders relevant ist und neuere Erkenntnisse zeigen, dass mehr MS als NBD-Läsionen der weißen Substanz zentrale Venen aufweisen, muss berücksichtigt werden, dass Patienten mit NBD im Allgemeinen weniger Läsionen der weißen Substanz aufweisen als Patienten mit MS, so dass die Verwendung der zentralen Vene zur Unterscheidung schwierig bleiben kann. Andere Probleme, die bei der Unterscheidung der beiden Entitäten hilfreich waren, sind in Tabelle 1 aufgeführt. Es ist wichtig zu beachten, dass die anfängliche Manifestation von NBD eine tumoröse Hirnläsion sein kann, die sowohl von einer tumorösen MS-Präsentation als auch von einem Hirntumor kaum zu unterscheiden ist, wobei letzteres eine wesentliche differentialdiagnostische Überlegung darstellt.30,31 Kliniker müssen sich bewusst sein, dass NBD und MS sich nicht gegenseitig ausschließen müssen. Tatsächlich erfüllt eine Gruppe von Patienten mit etablierter NBD auch die diagnostischen Kriterien für MS und weist klinische, bildgebende und Labormerkmale (oligoklonale Banden im Liquor) von MS auf .32
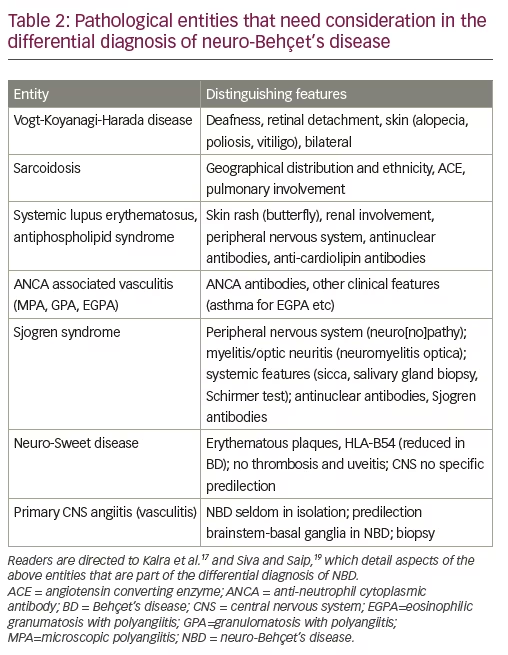
Ein weiterer wichtiger Aspekt der Differentialdiagnose ist die systemische Vaskulitis, die primär oder sekundär zu anderen systemischen Entzündungserkrankungen mit neurologischen Manifestationen sein kann. Tabelle 2 enthält eine Liste systemischer entzündlicher Erkrankungen mit oder ohne sekundäre Vaskulitis, die als NBD fehldiagnostiziert werden können, und ihre Unterscheidungsmerkmale, die bei der Differentialdiagnose von NBD hilfreich sein können. Diese werden in Kalra et al. und Siva und Saip.18,24
Bei Patienten mit akutem Schlaganfall ist die vaskuläre Variante der NBD manchmal Teil der Differentialdiagnose. Zur Unterscheidung von atherosklerotischen Schlaganfällen ohne Vaskulitis ist es hilfreich, die höhere Häufigkeit von Venenthrombosen im Vergleich zu arteriellen arteriellen Schlaganfällen bei NBD zu berücksichtigen. Auf der anderen Seite beinhalten einige Schlaganfälle bei NBD arterielle Hypertonie, so dass ein arterieller Schlaganfall eine Manifestation von NBD sein kann. Es ist auch wichtig, sich daran zu erinnern, dass Patienten mit BD ein höheres Risiko für kardiovaskuläre Ereignisse einschließlich Schlaganfall haben, unabhängig davon, ob sie NBD haben.33 Arterielle Dissektionen und Blutungen, wenn auch relativ selten bei NBD, sollten im entsprechenden klinischen Umfeld einen Verdacht darauf auslösen.
Psychiatrische NBD ist eine seltene Manifestation, normalerweise mit einer subakuten, progressiven Präsentation, und weist sehr unterschiedliche Manifestationen auf, die primäre degenerative Demenz, Depression oder offene Psychose nachahmen. Es ist sehr wichtig, NBD bei solchen Patienten zu berücksichtigen.34
Ein wichtiger Aspekt der Differentialdiagnose ist die Entwicklung eines neurologischen Problems bei einem Patienten mit etablierter BD. Die Kliniker müssen feststellen, ob das Problem mit NBD zusammenhängt oder nicht. Neben Schlaganfall sind Kopfschmerzen eine sehr häufige Beschwerde bei Menschen mit BD und stellen im Allgemeinen keine Manifestation von NBD dar. Wachsamkeit ist jedoch erforderlich, da Kopfschmerzen oft andere Manifestationen von NBD ankündigen.
Periphere Neuropathie tritt selten als klinische Manifestation von NBD auf, und wenn sie bei einer Person mit BD und ohne andere neurologische Manifestationen diagnostiziert wird, ist eine Suche nach alternativen Ätiologien gerechtfertigt.35
Schließlich betrifft ein aufkommender differentialdiagnostischer Aspekt neurologische Komplikationen der BD-Behandlung. Es ist wichtig, solche Komplikationen nicht mit der Entwicklung von NBD zu verwechseln. Die Rolle von Cyclosporin bei NBD ist wichtig, da es konsequent mit einer möglichen Beschleunigung und Verschlechterung von NBD in Verbindung gebracht wurde. Andere Behandlungen können auch Komplikationen haben. Immunsuppressiva können gelegentlich Meningitis erleichtern, die für NBD verwechselt werden kann. Die Verwendung von Anti-TNF-Wirkstoffen wie Infliximab, von denen gezeigt wurde, dass sie bei NBD erfolgreich sind,36 sollte Wachsamkeit erfordern, da Demyelinisierung und andere neurologische Komplikationen mit diesen Wirkstoffen berichtet wurden, obwohl noch keine berichtet wurde bei Verwendung bei BD.
Thalidomid, das gelegentlich für BD verwendet wird, verursacht vorhersehbar eine Neuropathie, die nicht mit NBD zusammenhängt. Das posteriore reversible Enzephalopathie-Syndrom wurde als Komplikation mehrerer Immuntherapien einschließlich Anti-TNF-Mitteln berichtet, und auf dem CONy-Symposium 2017 wurde ein interessanter Fall bei BD von Dr. George Vavougios aus Griechenland berichtet (Mündliche Kommunikation, CONy-Kongress, Athen 2017).
Zusammenfassend ist NBD eine schwerwiegende neuroinflammatorische Erkrankung, die große diagnostische, differentialdiagnostische und therapeutische Herausforderungen mit sich bringt. Das Bewusstsein für seine klinischen Merkmale und therapeutischen Möglichkeiten sowie die frühzeitige Diagnose sind für das Management von NBD von wesentlicher Bedeutung.