Das häufigste klinische Erscheinungsbild des primären Hyperparathyreoidismus (PHP) ist asymptomatische Hyperkalzämie, und die Diagnose von PHP basierend auf dem Vorhandensein von Knochenmanifestationen wie Osteitis fibrosa cystica (OFC) wird immer seltener. OFC tritt bei weniger als 5% der Patienten mit PHP auf und deutet auf eine schwerere oder lang anhaltende Erkrankung hin. OFC ist gekennzeichnet durch das Auftreten von Knochenschmerzen, die mit dem Auffinden spezifischer radiologischer Veränderungen verbunden sind, wie z. B. einer erhöhten subperiostalen Knochenresorption im distalen Drittel des Radius und der Mittelphalangen, einer Ausdünnung des distalen Schlüsselbeins, eines „Salz- und Pfefferschädels“, Knochenzysten und brauner Tumoren in langen Knochen. Braune Tumoren resultieren aus Knochendemineralisierung mit Osteoklastenaktivierung, Mikroblutungen und Mikrofrakturen und werden aufgrund ihrer typischen Farbe aufgrund reichlicher Hämosiderinablagerungen so genannt. Histopathologisch besteht eine Kombination von osteoklastischer und osteoblastischer Aktivität mit Zystenbildung und vielen Hämosiderin-beladenen Makrophagen.1 Die Differentialdiagnose von Brauntumoren umfasst das reparative Riesenzellgranulom und den Riesenzelltumor (GCT) des Knochens.
Der Fall eines Patienten mit PHP aufgrund eines Nebenschilddrüsenadenoms mit braunen Tumoren, die eine metastasierte GCT nachahmen, wird berichtet.
Ein 47-Jähriger besuchte im Mai 2008 zum ersten Mal die Abteilung für orthopädische Chirurgie eines Krankenhauses in Kastilien-La Mancha und klagte über Schmerzen in der Hüfte und der linken Hand, die nicht mit einem früheren Trauma verbunden waren. Der Patient berichtete über eine persönliche Vorgeschichte von Dyslipidämie, Typ-2-Diabetes mellitus, arterieller Hypertonie, Fettleibigkeit Grad I und Nierenkolik mit Calciumoxalatsteinen. Seine Familiengeschichte umfasste zwei Töchter, die wegen eines Adenoms diagnostiziert und operiert worden waren. Eine einfache Röntgenaufnahme der Hüfte und der Hand zeigte ein polylobuliertes zystisches Bild, das den kortikalen Knochen im dritten linken Mittelhandknochen und die supra-acetabulären und linken ilioischiopubischen Ramus-lytischen Läsionen aufblähte und verdünnte. Ein CT-Scan des Beckens (November 2008) zeigte große Läsionen in der Iliaca Ala, dem Ischium, dem linken Schambein und dem rechten Sakralflügel und Schenkelhals. Basierend auf diesen Befunden wurde der Patient im Juli und September 2009 operiert, bestehend aus Kürettage und Füllung sowohl mit einem autologen Transplantat als auch mit Knochenersatz des dritten linken Mittelhandknochens und der linken supra-acetabulären Läsion. Das pathologische Labor meldete eine GCT. Nachfolgende Kontrollen mit CT und MRT von Brust und Becken zeigten die Vergrößerung polylobulierter und expansiver lytischer Läsionen im Becken (Abb. 1), Kreuzbein, rechter Schenkelhals und L5 mit dem Auftreten einer neuen Läsion im rechten Femurkopf und im linken siebten Rippenbogen (Abb. 2). Diese Veränderungen wurden auf das Fortschreiten des metastasierten Tumors zurückgeführt. Aufgrund anhaltender Schmerzen, die das Gehen vollständig verhinderten, wurde der Patient an die Knochentumoreinheit des Hospital Universitario La Paz überwiesen, wo eine Überprüfung pathologischer Proben zu dem Schluss führte, dass die Knochenläsionen stark auf reparative Riesenzellgranulome hindeuteten und histologisch nicht von braunen Tumoren zu unterscheiden waren. Hyperparathyreoidismus wurde daher ausgeschlossen. Im November 2010 wurde der Patient an die endokrinologische Abteilung überwiesen, wo zusätzliche Labortests die folgenden Ergebnisse lieferten: Gesamtcalcium 14 mg / dl, korrigiertes Calcium 13,2 mg / dl, ionisches Calcium 1,72 mmol / l, Phosphat 1,9 mg / dl, Magnesium 1,86 mg / dL, Calcium im Urin 968,60 mg / 24h, Kreatinin 0,55 mg / dl, iPTH 535pg / ml, Vitamin D 13ng / ml. Die Ganzkörper-CT zeigte einen Knoten mit einem Durchmesser von 1,5 cm an der theoretischen Stelle der rechten Nebenschilddrüse, und ein Nebenschilddrüsen-Scan mit 20 MCI TC 99-Sestamibi ergab Befunde, die mit dem rechten hyperfunktionellen Nebenschilddrüsenadenom übereinstimmten. Darüber hinaus wurde das Vorhandensein eines assoziierten Phäochromozytoms sowohl aufgrund der Biochemie als auch der Morphologie ausgeschlossen. Es wurde eine rechte Parathyreoidektomie durchgeführt und eine intraoperative Biopsie als Nebenschilddrüsenadenom gemeldet. Die abschließende histopathologische Studie bestätigte das Vorhandensein eines Nebenschilddrüsenadenoms mit einem Gewicht von 4,5 g und einer Größe von 2,2 cm × 2 cm × 1,9 cm.
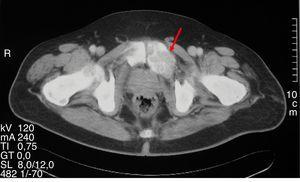
Abbildung 1.
Expansive lytische Läsion entsprechend Osteitis fibrosa cystica in einem Becken-CT-Scan.
(0.14 MB).
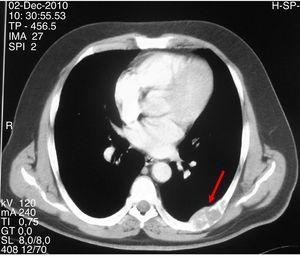
Abbildung 2.
CT-Scan, der eine expansive lytische Läsion mit gelapptem Umriss und kortikaler Ausdünnung im siebten linken Rippenbogen zeigt.
(0.21 MB).
Nach der Operation erlitt der Patient eine symptomatische Hypokalzämie, die eine Behandlung mit Calcitriol und Kalzium erforderte, die bis heute fortgesetzt wurde. Er wird weiterhin in der endokrinologischen Abteilung nachverfolgt, berichtet von einer signifikanten symptomatischen Besserung und kann mit Krücken gehen. Eine genetische Studie ergab keine Mutationen im MEN-1-Gen.
GCT ist ein hoch vaskularisierter Tumor, der in den Metaphysen oder Epiphysen langer Knochen oder im Becken, Kreuzbein oder Wirbel gefunden wird.2 Das radiologische und histologische Erscheinungsbild von braunen Tumoren, die für OFC typisch sind, kann einer GCT, wie sie bei unserem Patienten aufgetreten ist, sehr ähnlich sein, und die Differenzierung sollte anhand der klinischen Anzeichen und Laborergebnisse (iPTH) erfolgen. Einige der Autoren3,4 haben Fälle von OFC berichtet, bei denen aufgrund klinischer Anzeichen und Röntgenbilder zunächst ein Verdacht auf eine sekundäre metastasierende Knochenerkrankung bestand. Bei unserem Patienten basierte die Diagnose eines metastasierten primären Knochentumors jedoch auf histologischen Befunden, während die Familienanamnese, die in den oben genannten Fällen nicht berichtet wurde, mit PHP übereinstimmte.
Andererseits wird bei Patienten mit PHP häufig ein Vitamin-D-Mangel festgestellt3,5 im Zusammenhang mit einer Verschlimmerung der biochemischen und phänotypischen Darstellung der Krankheit (höhere Serum-PTH-Spiegel, große Nebenschilddrüsenadenome und höheres Frakturrisiko), was möglicherweise zum blumigen Krankheitsbild unseres Patienten beigetragen hat.
Es ist bekannt, dass familiäre Formen des Hyperparathyreoidismus selten sind (5%), und ihre häufigsten Ursachen sind multiple endokrine Neoplasien (MÄNNER) Typ 1 und 2A Syndrome, Hyperparathyreoidismus-Kiefertumor (HPT-JT) Syndrom und familiärer isolierter Hyperparathyreoidismus (FIHP).6 Bei MÄNNERN 1 ist Hyperparathyreoidismus die früheste und häufigste Darstellung (>90%), während er bei MÄNNERN 2A spät auftritt und eine geringe Penetranz aufweist. Obwohl die genetische Studie für MEN 1 bei unserem Patienten negativ war, sollte beachtet werden, dass in bis zu 30% der getesteten Fälle ein falsch negatives Ergebnis als Ergebnis von Mutationsmustern auftreten kann, die verschiedene Genregionen oder Mutationen in noch unbekannten Genen betreffen, die die Menin-Transkription oder -wirkung beeinflussen.7 Dies, zusammen mit dem wahrscheinlichen asynchronen Auftreten verschiedener Aspekte von MEN-1, macht eine kontinuierliche Überwachung notwendig. MEN 2A ist unwahrscheinlich, wenn keine Schilddrüsen-Neoplasie oder kein Phäochromozytom vorliegt. Differentialdiagnose sollte auch HPT-JT wegen des Ausmaßes der Knochenbefall und der großen Größe des Adenoms umfassen. Der endgültige Befund eines Nebenschilddrüsenkarzinoms hätte diese Diagnose aufgrund seines häufigen Auftretens bei HPT-JT unterstützt.8 Das Fehlen von Unterkiefer- oder Oberkieferfibro-ossären Läsionen und Nierenläsionen machte dies jedoch unwahrscheinlich. Obwohl FIHP in einigen Fällen eine Variante anderer hyperparathyreoider Syndrome darstellen kann, kann die Möglichkeit nicht ausgeschlossen werden, dass Mutationen, die sich an noch nicht identifizierten Orten befinden, die nicht bei MÄNNERN 1 und 2 und bei HPT-JT gemeldet wurden, dieses Syndrom verursachen können.
Das Interesse des gemeldeten Falles liegt in der Tatsache, dass er die Bedeutung der Beurteilung des Phosphor- und Kalziumstoffwechsels und der Nebenschilddrüsenfunktion bei allen Patienten mit Knochenläsionen, des Verdachts auf eine mögliche Ursache, wenn suggestive Läsionen vorliegen, und der Suche nach einer wahrscheinlichen zugrunde liegenden genetischen Komponente durch eine detaillierte familien- und persönliche Geschichte.