Wir sammelten Früchte von L. barbarum und L. ruthenicum in fünf Entwicklungsstadien, von jungen Früchten (≈10 Tage nach der Blüte) bis zu reifen (reifen) Früchten (34-45 Tage nach der Blüte), und untersuchten ihr Transkriptom und Metabolom.
- RNA-seq de novo assembly and functional annotation of unigenes
- Lycium barbarum (LB): paarweise Interstage-Analysen
- LB-Transkriptom
- LB Metabolom
- Lycium ruthenicum (LR): paarweise Interstage-Analysen
- LR-Transkriptom
- LR-Metabolom
- Interspezifische vergleichende Analyse von DEGs in verschiedenen Entwicklungsstadien
- Gesamt-DEGs während der Fruchtentwicklung
- Transkriptom – Pathways
- Transkriptomindividuelle Gene
- Metabolome – pathways
- Metabolom – individuelle Metaboliten
RNA-seq de novo assembly and functional annotation of unigenes
Wir haben insgesamt 30 cDNA-Bibliotheken aus Früchten von L. barbarum und L. ruthenicum mit drei biologischen Replikaten (drei Früchten von drei Bäumen) zu jedem Zeitpunkt vorbereitet: 2 Arten × 5 Zeitpunkte × 3 biologische Replikate. Die Proben wurden mit LB / LR (1-5) – (1-3) gekennzeichnet, wobei LB L. barbarum und LR L. ruthenicum ist, 1-5 Entwicklungsstadien von Früchten (S1–S5) und 1-3 Einzelproben (biologische Replikate) sind; so steht beispielsweise LB1-1 für L. barbarum, 1. beprobtes Entwicklungsstadium (S1), Fruchtprobe Nr.1 (von drei). Wir haben für diese 30 cDNA-Bibliotheken über 1,72 Milliarden paarweise Lesevorgänge generiert, was einem Durchschnitt von 57,2 Millionen Lesevorgängen pro Probe entspricht (ergänzender Datensatz S1). Eine strenge Qualitätsbewertung und Datenfilterung ergab insgesamt 801.766 hochwertige Lesevorgänge mit einer durchschnittlichen Länge von 730 und N50 von 1107 bp (Tabelle 1). Schließlich wurden aus den Transkripten insgesamt 326.276 Unigene mit der mittleren Länge von 596 bp und N50 von 847 bp erhalten (Tabelle 1). Korrelationskoeffizienten für RNA-seq-Daten für die 30 Proben weisen auf eine sehr gute Konsistenz der Ergebnisse zwischen biologischen Replikaten hin (Abb. 2).

Heatmap der Korrelationskoeffizienten für RNA-seq-Daten für 30 Proben von L. barbarum (LB) und L. ruthenicum (LR) Früchte in fünf verschiedenen Entwicklungsstadien. Die Proben sind mit LB/R1–5_1–3 gekennzeichnet, wobei LB L. barbarum, LR L ist. ruthenicum, 1-5 sind Fruchtentwicklungsstadien und 1-3 einzelne Proben. Die Proben wurden nach hierarchischem Clustering gruppiert; Dendrogramme oben und links von der Heatmap zeigen die Verwandtschaft der Proben an.
Von allen 326.276 Unigenen, die in öffentlichen Datenbanken abgefragt wurden, stimmten insgesamt 193.021 (59,15%) mit Genen und / oder Proteinen in mindestens einer Datenbank überein, und 12.171 (3,73%) wurden in allen Datenbanken annotiert. Die größte Anzahl von Unigenen (149.863, 45,93%) wurde in der NT-Datenbank und die niedrigste Anzahl (24.017; 7,36%) in der KOG-Datenbank annotiert.
Lycium barbarum (LB): paarweise Interstage-Analysen
LB-Transkriptom
Die höchste Anzahl von DEGs wurde in allen paarweisen Vergleichen der 1. Stufe und im Vergleich der 2. vs. 5. Stufe identifiziert (alle > 10.000 DEGs; Tabelle 2). Die kleinsten Zahlen wurden in 3rd vs. 4th und 4th vs. 5th stage Vergleiche (255-257) identifiziert. Die Heatmap-Analyse von DEGs in LB zeigt, dass ziemlich unterschiedliche Sätze von Genen in den frühen Entwicklungsstadien (1 + 2) und in späteren Stadien (3 bis 5) stark hochreguliert waren (Abb. 3A). Die Analyse der Probenbezogenheit zeigt, dass die Proben in zwei Kladen unterteilt werden konnten (Stufen 1 + 2 und 3 + 4 + 5 ), wobei die letztere Clade weiter in zwei Clades unterteilt ist: Stufen 3 + 4 und Stufe 5. Die intraspezifische KEGG-Funktionsklassifizierungsanalyse dieser DEGs identifizierte 15 Pathways signifikant (P < 0,05) differentiell reguliert zwischen verschiedenen Entwicklungsstadien (ergänzender Datensatz S2). Besonders stark differentiell reguliert waren ‚Pflanzenhormonsignaltransduktion‘, ‚Phenylpropanoidbiosynthese (b.)‘, ‚Linolsäurestoffwechsel (m.)‘, ‚Stärke und Saccharose m.‘, und ‚zeatin b.‘ (Fig. 3A).

Heatmaps und funktionelle Signalweganalysen von differentiell exprimierten Genen (DEGs) in Früchten von Lycium barbarum (Panel A) und L. ruthenicum (Panel B). Heatmaps wurden durch eine hierarchische Analyse von DEGs (y-Achse) und einzelnen Proben (x-Achse) generiert, wobei Dendrogramme über und links der Heatmap die Verwandtschaft von Proben anzeigen. Die Proben sind mit LB / R_1–5_1–3 gekennzeichnet, wobei auf das Artkürzel (LB oder LR) das Entwicklungsstadium der Frucht (1-5) und die Probennummer (1-3) folgen. Rechts neben den Heatmaps sind intraspezifische KEGG-Signalweganalysen von DEGs in allen fünf Entwicklungsstadien der beiden Arten dargestellt. Es werden nur die 15 besten angereicherten Pfade aufgelistet. q-Wert ist ein FDR-angepasster p-Wert.
Für eine eingehendere Analyse der Daten konzentrierten wir uns auf den Vergleich der am stärksten regulierten Signalwege in aufeinanderfolgenden Entwicklungsstadien. Im ersten paarweisen Vergleich (S1 vs S2) war ‚Phenylpropanoid b.‘ der am stärksten differentiell regulierte Weg, gefolgt von ‚Stärke und Saccharose m.‘ (Abb. 4). In beiden Signalwegen wurde eine sehr große Anzahl von DEGs (> 100) identifiziert. Ein ähnliches Ergebnis wurde im folgenden paarweisen Vergleich S2 vs. S3 beobachtet, jedoch trotz der relativ großen Anzahl von DEGs (>80) ‚Stärke und Saccharose m.‘ zeigte einen etwas niedrigeren q-Wert. Im Vergleich zwischen S3 und S4 war die Kohlenstofffixierung in photosynthetischen Organismen der am stärksten regulierte Weg, aber die Anzahl der Gene war viel geringer. Im letzten Paar, S4 vs. S5, ‚Zeatin b.‘, Flavonoid b.‘, Fettsäure b.‘ und ‚Galactose m.‘ waren die am stärksten regulierten Wege, aber keiner der Wege zeigte mehr als zwei DEGs.

LB transkriptom: pairwise interstage KEGG metabolic pathway enrichment. (A) Stufe 1 gegen Stufe 2. Stufe 2 vs. stufe 3. (C) Stufe 3 gegen Stufe 4. Stufe 4 vs. Stufe 5. Top 15 (oder alle, wenn insgesamt < 15) signifikant angereicherte Pfade werden angezeigt. Die Farbe des Balkens repräsentiert die Größe des q-Wertes (ein FDR-angepasster p-Wert). Farbkarte ist in der Abbildung dargestellt.
LB Metabolom
Wir führten paarweise Entwicklungsstadiumsvergleiche durch, um die Anreicherung von Metaboliten zwischen allen Stufenpaaren zu identifizieren. Die Anzahl der differentiell regulierten Metaboliten in paarweisen Vergleichen von Entwicklungsstadien in LB variierte von 66 (Stadien 3 vs. 4) bis 129 (Stadien 2 vs. 5). Die größte Anzahl in aufeinanderfolgenden Stufenvergleichen wurde zwischen der 1. und 2. Stufe beobachtet (117). Diese wurden einer großen Anzahl von Signalwegen zugeordnet; Die größte Anzahl von Metaboliten wurde ‚b. von Sekundärmetaboliten‘ (auch dem niedrigsten p-Wert) zugeordnet, gefolgt von ‚Proteinverdauung und -absorption‘ und ‚b. von Aminosäuren‘ (Abb. 5; ergänzende Abbildungen). In der aufeinanderfolgenden Stufe Paarvergleich (S2 vs. S3), ‚ABC-Transporter‘ war der am stärksten angereicherte Weg, gefolgt von ‚Purin m.‘. Im S3 vs. S4-Paarvergleich war ‚mikrobielles m. in verschiedenen Umgebungen‘ der am deutlichsten angereicherte Weg, gefolgt von ‚Carbapenem m.‘. Im S4 vs. S5-Paarvergleich zeigte eine relativ große Anzahl von Signalwegen ähnliche Ergebnisse (zwei Metaboliten und ähnliche p-Werte), aber bemerkenswert ist das Auftreten von ‚Isoflavonoid b.‘ und ‚Flavonoid b.‘ unter ihnen (Abb. 5; ergänzende Abbildungen).

LB metabolomische Daten: paarweise Anreicherung des KEGG-Stoffwechselwegs zwischen den Stadien. (A) Stufe 1 gegen Stufe 2. (B) Stufe 2 gegen Stufe 3. (C) Stufe 3 gegen Stufe 4. Stufe 4 vs. Stufe 5. Dieser Faktor ist das Verhältnis der Anzahl der signifikant regulierten Metaboliten im Stoffwechselweg und der Gesamtzahl der in diesem Stoffwechselweg annotierten Metaboliten (Bereich = 0 bis 1,0). Die Größe des Punktes repräsentiert die Anzahl der signifikant angereicherten Metaboliten im entsprechenden Weg, und die Farbe des Punktes repräsentiert den P-Wert (beide Legenden in der Abbildung gezeigt).
In Bezug auf einzelne Metaboliten wurde die Liste der im S1-Vergleich hochregulierten Metaboliten im S2-Vergleich von Trehalose, Glactinol und L-Äpfelsäure (alle ≈20 log2FC) angeführt. Ölsäure, 2-Oxoadipinsäure und Stearinsäure waren die am stärksten hochregulierten Metaboliten in S2 (alle ≈15 bis 18 log2FC). Die Liste der am stärksten hochregulierten Metaboliten in S2 im Vergleich zu S3 wurde von Dihydroxyaceton, LysoPC (18: 1 (9Z)) und Adenin (alle ≈16 bis 17,5 log2FC) angeführt. Die Liste der am stärksten hochregulierten Metaboliten im S3 (im Vergleich zu S2) wurde von Trehalose, Galactinol und L-Apfelsäure (alle ≈19 bis 20 log2FC) angeführt. Im Vergleich zwischen S3 und S4 wurde die Liste der am stärksten hochregulierten Metaboliten im S3 von L-Apfelsäure, DL-Arginin und Ölsäure (alle ≈18 bis 19 log2FC) angeführt, während die Liste der am stärksten hochregulierten Metaboliten im S4 von D-Mannose, N-Acetyllactosamin und LysoPC (18: 1 (9Z)) in LB (alle ≈16 log2FC) angeführt wurde. In der S4 vs. S5 Vergleich, die Liste der am stärksten hochregulierten Metaboliten in der S4 wurde von Trehalose, Palmitinsäure, N-Acetyllactosamin (alle ≈17 bis 18 log2FC) in LB gekrönt. Die Liste der am stärksten hochregulierten Metaboliten im S5 wurde von L-Norleucin, Anthranilsäure (Vitamin L1) und DL-Arginin (alle ≈16 bis 18 log2FC) angeführt (ergänzender Datensatz S3).
Lycium ruthenicum (LR): paarweise Interstage-Analysen
LR-Transkriptom
Die Heatmap-Analyse von DEGs in LR zeigt, dass in den frühen Entwicklungsstadien (1 + 2) und in reifen Früchten (Stadium 5) fast völlig unterschiedliche Gensätze stark hochreguliert waren, wobei nach der zweiten Stufe ein offensichtlicher transkriptomischer Reset auftrat (Abb. 3B). Die Analyse der Probenbezogenheit zeigt, dass die Proben in zwei Kladen unterteilt werden konnten (Stufen 1 + 2 und 3 + 4 + 5 ), wobei die letztere Clade weiter in zwei Clades unterteilt ist: Stufen 3 + 4 und Stufe 5. Die intraspezifische KEGG-Funktionsklassifizierungsanalyse dieser DEGs identifizierte 35 Pathways signifikant (P < 0,05) differentiell reguliert zwischen verschiedenen Entwicklungsstadien (ergänzender Datensatz S2). Besonders stark differentiell reguliert waren ‚Photosynthese‘, ‚Linolsäure m.‘, ‚Isochinolinalkaloid b.‘, ‚Flavonoid b.‘ und ‚Stärke und Saccharose m‘ (Abb. 3B). Die höchste Anzahl von DEGs wurde in den meisten paarweisen Vergleichen der Stufe 1 (1 vs. 2 war eine Ausnahme) und im Vergleich der Stufe 2 vs. 5 (alle > 5.000 DEGs; Tabelle 2) identifiziert. Die mit Abstand kleinste Zahl wurde im Vergleich S3 vs. S4 identifiziert (39). Im S1 vs S2 paarweisen Vergleich war ‚Aminozucker und Nukleotidzucker m.‘ der am stärksten differentiell regulierte Weg, gefolgt von ‚Linolsäure m.‘ (Abb. 6). Im paarweisen Vergleich von S2 und S3 waren die am deutlichsten differentiell regulierten Signalwege ‚Isochinolinalkaloid b.‘, ‚Tyrosin m.‘, ‚Phenylpropanoid b‘ und ‚Flavonoid b.‘. In beiden paarweisen Vergleichen wurde die höchste Anzahl von DEGs (> 15 bzw. > 35) in der Stärke- und Saccharose-Tabelle identifiziert. In der S3 vs. S4 Vergleich, ‚(Alpha-) Linolsäure m.‘ war der am stärksten regulierte Weg, aber die Anzahl der Gene war viel niedriger. Im Vergleich zwischen S4 und S5 waren ‚Phenylpropanoid b‘ (auch die größte Anzahl von DEGs) und ‚Linolsäure m.‘ die am stärksten regulierten Wege.
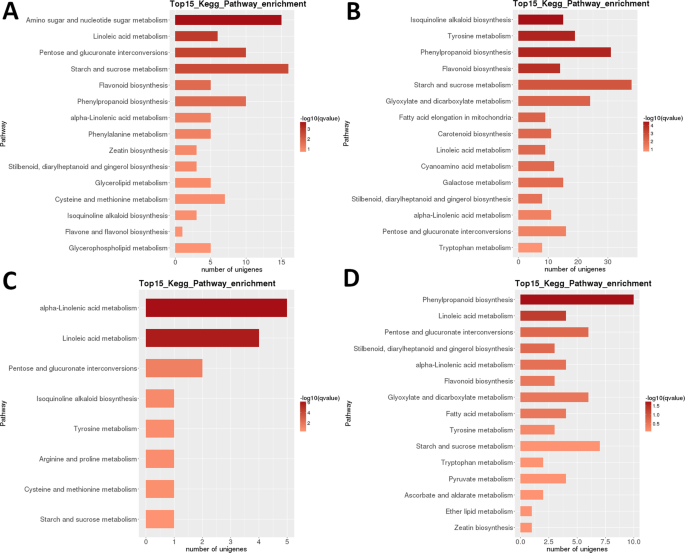
LR transkriptom: pairwise interstage KEGG metabolic pathway enrichment. (A) Stufe 1 gegen Stufe 2. (B) Stufe 2 gegen Stufe 3. (C) Stufe 3 gegen Stufe 4. Stufe 4 vs. Stufe 5. Top 15 (oder alle, wenn insgesamt < 15) signifikant angereicherte Pfade werden angezeigt. Die Farbe des Balkens stellt die Größe des q-Wertes dar (ein FDR-angepasster p-Wert, Farbkarte in der Abbildung).
LR-Metabolom
Die Anzahl der differentiell regulierten Metaboliten in paarweisen Vergleichen von Entwicklungsstadien in LR variierte von 66 (Stadien 3 vs. 4) bis 133 (Stadien 2 vs. 5) (Ergänzender Datensatz S3). In aufeinanderfolgenden Stufenvergleichen wurde die größte Anzahl zwischen der 1. und 2. Stufe beobachtet (117). Diese wurden einer großen Anzahl von Pfaden zugeordnet; mit der größten Anzahl von Metaboliten, die ‚b von Sekundärmetaboliten‘ zugeordnet sind, gefolgt von ‚Proteinverdauung und -absorption‘, ‚b. von Aminosäuren‘ und ‚Flavonoid b‘. Im Vergleich zwischen S2 und S3 war ‚ABC-Transporter‘ der am stärksten angereicherte Signalweg, gefolgt von ‚Purin m‘. Im S3 vs. S4-Paarvergleich war ‚mikrobielles m. in verschiedenen Umgebungen‘ der am deutlichsten angereicherte Weg, gefolgt von ‚Carbapenem m.‘. In der S4 vs. S5 Im Vergleich zeigte eine relativ große Anzahl von Signalwegen ähnliche Ergebnisse (2 Metaboliten und ähnliche p-Werte), aber bemerkenswert ist das Auftreten von ‚Isoflavonoid b.‘ und ‚Flavonoid b.‘ unter ihnen (Abb. 7; ergänzende Abbildungen).

LR metabolomische Daten: paarweise Anreicherung des KEGG-Stoffwechselwegs zwischen den Stadien. (A) Stufe 1 gegen Stufe 2. (B) Stufe 2 gegen Stufe 3. (C) Stufe 3 gegen Stufe 4. Stufe 4 vs. Stufe 5. Dieser Faktor ist das Verhältnis der Anzahl der signifikant regulierten Metaboliten im Stoffwechselweg und der Gesamtzahl der in diesem Stoffwechselweg annotierten Metaboliten (Bereich = 0 bis 1,0). Die Größe des Punktes repräsentiert die Anzahl der signifikant angereicherten Metaboliten im entsprechenden Weg, und die Farbe des Punktes repräsentiert den P-Wert (beide Legenden in der Abbildung gezeigt).
In Bezug auf einzelne Metaboliten (Ergänzungsdatensatz S3), in der S1 vs. Im Vergleich zu S2 wurde die Liste der in S1 hochregulierten Metaboliten von Trehalose, Galactinol und L-Äpfelsäure (≈19-21 log2FC) angeführt, während Ölsäure, 2-Oxoadipinsäure und Stearinsäure die am stärksten hochregulierten Metaboliten in S2 waren (≈15-18 log2FC). Im Vergleich zwischen S2 und S3 waren Dihydroxyaceton, Indoxylsulfat und N-Acetyllactosamin die am stärksten hochregulierten Metaboliten im S2 (≈17-19,5 log2FC) und Trehalose, Galactinol und L-Äpfelsäure (≈19-21 log2FC) im S3. In der S3 vs. S4-Vergleich, L-Äpfelsäure, DL-Arginin und Ölsäure wurden im S3 (≈16-19 log2FC) und 1,7-Dimethylxanthin, D-Mannose und N-Acetyllactosamin (≈ 15-17 log2FC) im S4 hochreguliert. Im Vergleich S4 vs. S5 wurden Flavinmononukleotid, Trehalose und Isoferulasäure im S4 hochreguliert (≈18-20 log2FC), während PG (16: 0/18: 1 (9Z)), D-Prolin und DL-Arginin am stärksten hochreguliert waren (alle ≈16 bis 18 log2FC) Metaboliten.
Interspezifische vergleichende Analyse von DEGs in verschiedenen Entwicklungsstadien
Gesamt-DEGs während der Fruchtentwicklung
Interspezifischer paarweiser Stadiumsvergleich (LR1 vs. LB1, LR2 vs.LB2, etc.) zeigt, dass 928 DEGs von allen fünf Paaren geteilt wurden (Abb. 8A). Die höchste Anzahl von DEGs wurde in Stufe 3 (3989) und die niedrigste in Stufe 4 (2825) identifiziert (Abb. 8B); während die höchste Anzahl von DEGs, die für ein Paar eindeutig sind, in den Stufen 3 (574), 1 und 5 (beide 554) und die niedrigste in Stufe 4 (126) beobachtet wurden (Abb. 8A). Die Anzahl der auf- und abgeregelten DEGs war in jedem der paarweisen Stufenvergleiche relativ ähnlich; z. B. wurden in Stufe 5 1668 DEGs hochreguliert und 1670 DEGs in LR im Vergleich zu LB herunterreguliert (Abb. 8B). In den anderen vier Stufen war die Anzahl der hochregulierten Gene jedoch etwas höher (93 bis 189 GRAd).

Gene differentiell exprimiert (DEGs) zwischen Früchten von L. barbarum (LB) und L. ruthenicum (LR). (A) Detaillierte stufenweise (1 bis 5) Vergleiche (LB vs. LR). (B) Die Anzahl der hochregulierten (rot) und herunterregulierten (grün) DEGs in LR im Vergleich zu LB in fünf untersuchten Entwicklungsstadien.
Transkriptom – Pathways
Die Heatmap-Analyse von DEGs zeigt, dass die Früchte zweier Arten in allen Entwicklungsstadien sehr unterschiedliche Genexpressionsprofile aufweisen, biologische Replikate jedoch sehr ähnliche Profile aufweisen, was auf eine begrenzte individuelle Variabilität in jedem Entwicklungsstadium hinweist (Ergänzende Abbildungen: Abb. S9). Die vergleichende Analyse der KEGG-Signalweganreicherung zeigt, dass nur einige Signalwege in LR im Vergleich zu LB in allen fünf Entwicklungsstadien konsistent hoch angereichert waren (in Bezug auf die Genregulation) (Abb. 9). Insbesondere die Signaltransduktion von Pflanzenhormonen (2.-höchste in S1, 8.-höchste in S2, die höchste in S3, S4 und S5) und die Interaktion zwischen Pflanzen und Pathogenen (die höchste in S1, 3.-höchste in S2, 2.-höchste in S3, 4.-höchste in S4 und 15.-höchste in S5) waren in allen Stadien relativ stark hochreguliert. Phenylpropanoid-Biosynthese (nicht in Top 15 in S1, die höchste in S2, 3. höchste in S3, 7. höchste in S4, 11. höchste in S5), Ubichinon und andere Terpenoid-Chinon-Biosynthese (nicht in Top 15 in S1, 6. höchste in S2, 7. höchste in S3, 2. höchste in S4, 6. höchste in S5) waren ebenfalls in allen Stadien mit Ausnahme der ersten relativ stark hochreguliert. Der Flavonoid-Biosyntheseweg war in frühen Stadien nicht hoch angereichert (nicht in Top 15 in S1, 14.-höchste in S2) und in späten Stadien hoch angereichert (3. bis 4.-höchste in den Stadien 3 bis 5). Der (alpha-) Linolsäurestoffwechsel war in mittleren Stadien stark angereichert (9.-höchste in S1, 2.-höchste in S2, 5. und 6.-höchste in S3, 8.-höchste in S4, nicht in Top 15 in S5).

Vergleichende Analyse der KEGG-Stoffwechselweganreicherung. Links (rot) sind die Top 15 der mit L. ruthenicum im Vergleich zu L. barbarum angereicherten Pfade und rechts (grün) die mit L. barbarum im Vergleich zu L. ruthenicum angereicherten Pfade dargestellt. Entwicklungsstadien (1-5) sind in der Abbildung angegeben. q-Wert ist ein FDR-angepasster p-Wert.
Unter den in L. ruthenicum im Vergleich zu L. barbarum herunterregulierten Pfaden (Abb. 9) es wurden bemerkenswerte Veränderungen zwischen den frühen Stadien (1 und 2) beobachtet, in denen der Cyanaminosäurestoffwechsel und die Carotinoidbiosynthese am stärksten herunterreguliert waren, und den späten Stadien (4 und 5), in denen SNARE-Wechselwirkungen beim vesikulären Transport, im Nikotinat- und Nikotinamidstoffwechsel sowie im Porphyrin- und Chlorophyllstoffwechsel durchweg relativ stark herunterreguliert waren.
Transkriptomindividuelle Gene
Unter den am stärksten differentiell exprimierten Genen waren einige entwicklungsstadienspezifisch (d. h. nur in frühen oder späten Entwicklungsstadien stark differentiell reguliert), andere jedoch in allen fünf untersuchten Stadien konsistent stark differentiell reguliert (Ergänzender Datensatz S4). Mehrere immunitätsbezogene Gene, die in LR im Vergleich zu LB in den frühen Entwicklungsstadien sehr hochreguliert sind, gehören zu den Beispielen für das entwicklungsstadienspezifische Expressionsmuster: chitinase war der 2. höchste hochregulierte GRAD in S1 (13,43-fach), der höchste in S2 (13,89-fach), aber in späteren Stadien war es kein GRAD. In ähnlicher Weise war der EIX-Rezeptor 1/2 in frühen Stadien ebenfalls sehr stark hochreguliert, am höchsten in S1 (13,70) und am 2. höchsten in S2 (10,90), wurde jedoch in späteren Stadien auch nicht als DEG identifiziert. Einige wachstumsbezogene Gene zeigten auch ein ähnliches Expressionsmuster: Phosphoglyceratkinase (PGK) gehörte zu den Wenigen am stärksten hochregulierten Genen in den ersten drei Stadien (13.14, 12.87 und 12.77 jeweils), aber es wurde auch nicht als DEG in späteren Stadien identifiziert. CCR4-NOT-Transkriptionskomplex-Untereinheit 7/8 (CNOT7 / 8) zeigte ebenfalls ein sehr ähnliches Expressionsmuster: in den ersten drei Stufen stark hochreguliert und in den Stufen 4 und 5 kein GRAD. Mehrere Flavonoid- und Phenylpropanoid-Biosynthese-assoziierte Gene zeigten ein umgekehrtes entwicklungsstadienspezifisches Expressionsmuster mit relativ geringer Expression in frühen Stadien und sehr hoch in späteren Stadien. Beispiele sind: bifunktionelle Dihydroflavonol-4-Reduktase/Flavanon-4-Reduktase (DFR), die in LR in S1 (2.25), kein GRAD in S2, hochreguliert in S3 (7.79) und 3.-höchstes hochreguliertes Gen in S4 (14.25) und S5 (16.03). Ein Paralog dieses Gens zeigte ein fast identisches Muster: leicht hochreguliert in S1 (2.44), kein Grad in S2, hoch hochreguliert in S3 (7.40), 6th-höchster hochregulierter GRAD in S4 (13.26) und 5th in S5 (14.59). In ähnlicher Weise war Flavonoid 3′, 5′-Hydroxylase (F3’5’H) in den ersten beiden Stadien kein GRAD, hochreguliert in S3 (6.69), 5th-höchstes hochreguliertes Gen in S4 (13.42) und 4th-höchste in S5 (15.05). Flavonoid-O-Methyltransferase (OMT) war kein GRAD in der S1, aber in S2 zeigte es bereits ein mittelhohes Hochregulationsniveau (4,32), von der S3 war es bereits die dritthöchste hochregulierte DEG (13,30), und es war das am höchsten hochregulierte Gen in S4 (18,73) und S5 (18,10). Leucoanthocyanidindioxygenase (LDOX; Anthocyanbiosynthese) war in S1 und S2 kein GRAD, gefolgt von einer hohen bis sehr hohen Hochregulierung in späteren Stadien (5.63, 9.44, 11.56). Zwei Chalkonsynthase-Paraloge (CHS und CHS2; Flavonoid-Biosynthese) waren auch in S1 und S2 nicht stark reguliert (CHS2: kein GRAD, CHS: -1.14 in S1, kein GRAD in S2), aber in S3–S5 zeigten beide Gene eine mittelhohe bis hohe Hochregulation (CHS2: 5.32, 7.84, 6.00; und CHS: 4.67, 7.01, 6.82; beziehungsweise). Wir haben diese Gene für die qPCR-Analyse ausgewählt, und die Ergebnisse stimmen sehr gut mit den RNA-seq-Daten überein (Ergänzende Ergebnisse; Ergänzender Datensatz S5). Schließlich wurde auch die Cytokinindehydrogenase, ein mit der Zeatin-Biosynthese verbundenes Gen, in den letzten drei Stadien zunehmend hochreguliert (2,6–5,7).
Einige Gene wurden jedoch in allen fünf untersuchten Stadien konsistent differentiell exprimiert. Beispiele umfassten auch einige immunitätsbezogene Gene, wie zwei Paralogue der Glutathion-S-Transferase, die in LR im Vergleich zu LB in allen Stadien hochreguliert waren: 9,38 und 8,58 (alle Werte als Faltänderungen in der jeweiligen Reihenfolge dargestellt) in S1, 6,30 und 6,34 in S2, die 2. und 7. höchsten hochregulierten DEGs in S3 (14,08 und 12,70), 2. und 4. höchste in S4 (15,71 und 14.16) und 2. und 6. höchste in S5 (16.48 und 14.40). Das Pflanzenkrankheitsresistenzprotein RPM1 war ebenfalls in allen fünf Stadien stark hochreguliert (S1 = 13,15; S2 = 12,08; S3 = 13,11; S4 = 12,81; S5 = 13:94). Unter den konsequent differentiell exprimierten Genen in allen Entwicklungsstadien befanden sich auch einige, die mit dem Aminosäurestoffwechsel in Verbindung standen, aber ihr Muster war im Vergleich zu früheren Beispielen umgekehrt: Sie zeigten eine hohe Herunterregulierung in LR im Vergleich zu LB. Beispiele sind Acetyl-CoA-Acyltransferase 1 (AAT1; Valin−, Leucin- und Isoleucinabbau) mit einem zeitlichen Profil zunehmend hoher Herunterregulation, beginnend von -7,0 in der S1 bis < -10-fach in den letzten drei Stufen. Prolin-Iminopeptidase, die mit dem Arginin- und Prolin-Metabolismus assoziiert ist, war in LR in allen Stadien stark herunterreguliert: S1 = -9,75, S2 = -10,89 (3. höchster Wert), S3 = -11,05 (4. höchster Wert), S4 = -10,01 und S5 = -11,98 (3. höchster Wert). Schließlich wurde die 5-Methyltetrahydropteroyltriglutamat–Homocystein-Methyltransferase (metE) in LR in allen Stadien durchweg extrem stark herunterreguliert: 2. höchste in S1 (-11,76), die höchste in S2 (-11,74), 3. höchste in S3 (-11,43), die höchste in S4 (-12,36) und 2. höchste in S5 (-12,83). Zwei DNA-Replikations- und transkriptionsassoziierte Gene waren in LR in allen Stadien ebenfalls stark herunterreguliert: das GTP-bindende Kernprotein Ran (RAN; -10,0 bis -12,0) und der Replikationsfaktor A1 (RFA1) (-8,0 bis -12,0). Einige wachstums- und stressbedingte Gene waren auch bei LR durchweg stark herunterreguliert: heterogenes nukleares Ribonukleoprotein A1 / A3 (hnRNP; -7 bis -11) und Hitzeschock 70 kDa Protein 1/8 (HSPA1_8) S1 = -4,95, S2 = -8,88, S3 = -11,48 (2. höchster Wert), S4 = -9,36, S5 = -12,89 (höchster Wert). Interessanterweise wurde auch ein mit der Phenylpropanoid-Biosynthese verbundenes Gen, die Shikimathydroxycinnamoyltransferase (HCT), in LR durchweg stark herunterreguliert: S1 = -6,82, S2 = -8,14, S3 = -11,71 (das höchste), S4 = -11,00 (3.-höchste), S5 = -11,91 (4.-höchste). Ein Schlüsselregulator der Anthocyanbiosynthese, der Transkriptionsfaktor MYB114, war jedoch in LR in allen fünf Entwicklungsstadien hochreguliert: 6.11, 4.69, 7.47, 9.05 und 8.95 (S1–S5).
Metabolome – pathways
Wir führten auch eine vergleichende interspezifische stufenweise Analyse von Stoffwechselwegen durch (Abb. 10). In der ersten Entwicklungsstufe (S1) identifizierten wir 39 differentiell regulierte Metaboliten. Unter den 20 wichtigsten Stoffwechselwegen, mit denen diese Metaboliten assoziiert waren, waren einige von ihnen mit Aminosäuren assoziiert, aber die Gesamtzahl der Metaboliten pro Stoffwechselweg war relativ gering (1-2), und die P-Werte deuteten nicht auf ein hohes Maß an Signifikanz hin (Abb. 10 – tafel 1). Insbesondere der Vitamin-B6-Metabolismus (m) und das mikrobielle m in verschiedenen Umgebungen zeigten beide vergleichsweise hohe P-Werte, einen Anreicherungsfaktor (EF) von 1,0 und 2 identifizierte Metaboliten. In der S2 identifizierten wir 58 differentiell regulierte Metaboliten, die mit nur vier Signalwegen assoziiert waren: Tryptophan m, Phenylpropanoid-Biosynthese (b), b von Phenylpropanoiden (dies sind zwei verschiedene Wege in der KEGG-Datenbank) und Phenylalanin, Tyrosin und Tryptophan b (alle EF = 1,0, 2-3 Metaboliten und p < 0,5; Abb. 10 – tafel 2). In der S3 identifizierten wir 59 differentiell regulierte Metaboliten, die mit 19 Signalwegen assoziiert waren, die meisten mit dem EF 1.0, aber relativ nicht signifikanten P-Werten (> 0.5; Abb. 10 – tafel 3). Wege mit einer relativ hohen Anzahl von Metaboliten (n = 5) waren: Proteinverdauung und -absorption, b von pflanzlichen Sekundärmetaboliten, b von Antibiotika und b von Aminosäuren. In der S4 identifizierten wir 58 differentiell regulierte Metaboliten, die mit einer großen Anzahl von Signalwegen, meist mit dem EF 1.0, und vergleichsweise hohen Signifikanzwerten (meist P > 0.5; Abb. 10 – tafel 4). Wege mit relativ hoher Anzahl von Metaboliten (n ≥ 3) waren: Phenylpropanoid b, Phenylalanin, Tyrosin und Tryptophan b, Glucosinat b, b von Alkaloiden, die vom Shikimat-Weg abgeleitet sind, und 2-Oxocarbonsäure m. In der reifen Frucht (S5) identifizierten wir 39 differentiell regulierte Metaboliten, die mit einer großen Anzahl von Wegen assoziiert waren, jedoch meist mit niedrigen P-Werten und nur 1 Metaboliten pro Weg (Abb. 10 – platte 5). Stoffwechselwege mit mehr als 1 Metabolit waren: proteinverdauung und -absorption, Phenylpropanoid b, Mineralabsorption, zentraler Kohlenstoff m bei Krebs, b von Sekundärmetaboliten, b von Phenylpropanoiden und Aminoacy-tRNA b. Die Hauptkomponentenanalyse (PCA) aller Daten (2 Arten × 5 Stadien × 5 biologische Replikate) ergab eine hohe Ähnlichkeit zwischen biologischen Replikaten (Clustering) und bestätigte eine bemerkenswerte Variabilität zwischen verschiedenen Fruchtreifestufen für beide Arten (Abb. 10 – tafel 6).

Vergleichende Analyse der KEGG-Stoffwechselweganreicherung. Links (rot) sind die Top 15 der mit L. ruthenicum im Vergleich zu L. barbarum angereicherten Pfade und rechts (grün) die mit L. barbarum im Vergleich zu L. ruthenicum angereicherten Pfade dargestellt. Entwicklungsstadien (1-5) sind in der Abbildung angegeben. q-Wert ist ein FDR-angepasster p-Wert.
Metabolom – individuelle Metaboliten
Die Liste (ergänzender Datensatz S6) der am stärksten differentiell regulierten Metaboliten zwischen den beiden Arten zeigte einige Unterschiede zwischen den fünf Fruchtentwicklungsstadien. Interessanterweise war Fructose-1-Phosphat in allen fünf Stadien der am stärksten hochregulierte Metabolit in LR im Vergleich zu LB: log2-Faltenänderung = 6,3, 7,6, 7,7, 8,1 und 6,5 (Stadien 1 bis 5). In Bezug auf die in LB hochregulierten Metaboliten gab es mehr Unterschiede zwischen den Stadien: in der S1 waren die Unterschiede eher gering, wobei 9-Decenol der am stärksten hochregulierte Metabolit war (log2-fache Veränderung = 2,7; im Vergleich zu LR). Analysen von S2 und S3 ergaben sehr kongruente Ergebnisse, wobei Phenol (3.7 bzw. 3.2) der am stärksten hochregulierte Metabolit war. In der S4 war Indoxylsulfat (4.7) der Top-Metabolit, der in LB hochreguliert wurde. Eine metabolische Verschiebung wurde in der reifen Frucht (S5) beobachtet, wo die Liste der in LB hochregulierten Metaboliten von Stearoylcarnitin (7.1), Methoxyessigsäure (5.3), S-Methyl-5′-thioadenosin (4.7), Lisinopril (4.7), Adenosin 3′,5′-cyclisches Phosphat (cAMP) (4.7) usw. Andere in LR hochregulierte Metaboliten (abgesehen von Fructose-1-phosphat) waren Naringin (6,2), Lauroyl-CoA (4,8), L-Phenylalanin (4,6) usw.