Recolectamos frutos de L. barbarum y L. ruthenicum en cinco etapas de desarrollo, desde frutas jóvenes (≈10 días después de la floración) hasta frutas maduras (maduras) (34-45 días después de la floración), y estudiamos su transcriptoma y metabolome.
- Ensamblaje de novo y anotación funcional de ARN-seq de unigenos
- Lycium barbarum( LB): análisis interetapa en pares
- Transcriptoma en PARES
- Metaboloma LB
- Lycium ruthenicum (LR): análisis de pares entre etapas
- Transcriptoma de LR
- Metaboloma LR
- Análisis comparativo interespecífico de DEGs en diferentes etapas de desarrollo
- DEGs totales durante el desarrollo de la fruta
- Las vías del transcriptoma
- Genes individuales del transcriptoma
- Vías metabólicas
- Metaboloma – metabolitos individuales
Ensamblaje de novo y anotación funcional de ARN-seq de unigenos
Preparamos un total de 30 bibliotecas de ADNc de frutos de L. barbarum y L. ruthenicum, con tres réplicas biológicas (tres frutos de tres árboles) en cada punto de tiempo: 2 especies × 5 puntos de tiempo × 3 réplicas biológicas. Las muestras fueron etiquetadas LB/LR (1-5)-(1-3), donde LB es L. barbarum y LR es L. ruthenicum, 1-5 son etapas de desarrollo de la fruta (S1–S5), y 1-3 son muestras individuales (réplicas biológicas); por ejemplo, LB1-1 representa L. barbarum, 1a etapa de desarrollo de la muestra (S1), muestra de fruta No.1 (de tres). Generamos más de 1,72 mil millones de lecturas de pares para estas 30 bibliotecas de ADNc, lo que corresponde a un promedio de 57,2 millones de lecturas por muestra (Conjunto de datos suplementario S1). La rigurosa evaluación de la calidad y el filtrado de datos arrojaron un total de 801.766 lecturas de alta calidad con una longitud media de 730 y un N50 de 1.107 pb (Tabla 1). Finalmente se obtuvo un total de 326.276 unigenes con una longitud media de 596 pa y N50 de 847 pa de las transcripciones (Tabla 1). Los coeficientes de correlación para los datos de ARN-seq de las 30 muestras indican una muy buena consistencia de los resultados entre las réplicas biológicas (Fig. 2).

Mapa de calor de coeficientes de correlación para datos de ARN-seq para 30 muestras de frutas de L. barbarum (LB) y L. ruthenicum (LR) en cinco etapas de desarrollo diferentes. Las muestras están etiquetadas con LB / R1–5_1-3, donde LB es L. barbarum, LR es L. ruthenicum, 1-5 son etapas de desarrollo de la fruta, y 1-3 muestras individuales. Las muestras se agruparon por agrupamiento jerárquico; los dendrogramas arriba y a la izquierda del mapa de calor indican la relación de las muestras.
De los 326.276 unigenes consultados en bases de datos públicas, un total de 193.021 (59,15%) genes y/o proteínas coincidentes en al menos una base de datos, y 12.171 (3,73%) fueron anotados en todas las bases de datos. El mayor número de unigenes (149.863, 45,93%) fue anotado en la base de datos NT, y el menor número (24.017, 7,36%) en la base de datos KOG.
Lycium barbarum( LB): análisis interetapa en pares
Transcriptoma en PARES
Se identificaron los números más altos de DEG en todas las comparaciones en pares de la 1ª etapa y en la comparación de la 2ª vs.la 5ª etapa (todas > 10.000 DEG; Tabla 2). Los números más pequeños se identificaron en comparaciones de las etapas 3ª vs. 4ª y 4ª vs. 5ª (255-257). El análisis de mapas de calor de DEGs en LB muestra que conjuntos de genes bastante diferentes estaban altamente regulados en las primeras etapas de desarrollo (1 + 2) y en etapas posteriores (3 a 5) (Fig. 3A). El análisis de la relación de la muestra indica que las muestras podrían dividirse en dos clados (etapas 1 + 2 y 3 + 4 + 5) , con el último clado subdividido en dos clados: etapas 3 + 4 y etapa 5. El análisis de clasificación funcional intraespecífica de KEGG de estos DEG identificó 15 vías de manera significativa (P < 0,05) reguladas de manera diferente entre diferentes etapas de desarrollo (Conjunto de datos suplementarios S2). La «transducción de señales de hormonas vegetales», la » biosíntesis de fenilpropanoides (b.)», el » metabolismo del ácido linoleico (m.)», el » almidón y sacarosa m.», y «zeatin b.» (Fig. 3A).

Mapas de calor y análisis de vías funcionales de genes expresados diferencialmente (DEGs) en frutas de Lycium barbarum (panel A) y L. ruthenicum (panel B). Los mapas de calor se generaron mediante un análisis jerárquico de DEGs (eje y) y muestras individuales (eje x), donde los dendrogramas por encima y por la izquierda del mapa de calor indican la relación de las muestras. Las muestras están etiquetadas con LB / R_1–5_1-3, donde el acrónimo de la especie (LB o LR) va seguido de la etapa de desarrollo de la fruta (1-5) y el número de muestra (1-3). Intraespecífica KEGG análisis de vía de los Grados en todas las cinco etapas de desarrollo en las dos especies se muestran a la derecha del mapa. Solo se enumeran las 15 principales vías enriquecidas. el valor q es un valor p ajustado por FDR.
Para un análisis más profundo de los datos, nos centramos en la comparación de las vías reguladas más significativas en etapas sucesivas de desarrollo. En la primera comparación de pares (S1 vs S2), el ‘fenilpropanoide b.’ fue la vía más regulada diferencialmente, seguida por ‘almidón y sacarosa m.’ (Fig. 4). Se identificaron un gran número de DEG (>100) en ambas vías. Un resultado similar se observó en la siguiente comparación de pares, S2 vs. S3, pero a pesar del número bastante grande de DEGs (>80) ‘almidón y sacarosa m.’exhibió un valor q algo menor. En la comparación S3 vs. S4, la’ fijación de carbono en organismos fotosintéticos ‘ fue la vía regulada más significativa, pero el número de genes fue mucho menor. En el último par, S4 vs. S5,’ zeatina b.’, flavonoide b.’, ácido graso b. ‘y’ galactosa m. ‘ fueron las vías reguladas más significativamente, pero ninguna de las vías exhibió más de dos DEG.

Transcriptoma LB: enriquecimiento de la vía metabólica de KEGG entre etapas por pares. (A) Etapa 1 vs.etapa 2. (B) Etapa 2 vs etapa 3. (C) Etapa 3 vs etapa 4. (D) Etapa 4 vs la etapa 5. Se muestran las 15 vías más enriquecidas (o todas si el total es <15). El color de la barra representa la magnitud del valor q (un valor p ajustado por FDR). La tabla de colores se muestra en la figura.
Metaboloma LB
Realizamos comparaciones de etapas de desarrollo por pares para identificar el enriquecimiento de metabolitos entre todos los pares de etapas. El número de metabolitos regulados diferencialmente en comparaciones de pares de estadios de desarrollo en LB varió de 66 (estadios 3 frente a 4) a 129 (estadios 2 frente a 5). El mayor número de comparaciones sucesivas de estadios se observó entre el 1º y el 2º estadio (117). Estos se asignaron a un gran número de vías; el mayor número de metabolitos se asignó a «b. de metabolitos secundarios» (también el valor p más bajo), seguido de «digestión y absorción de proteínas» y «b. de aminoácidos» (Fig. 5; Cifras Complementarias). En la fase sucesiva par de comparación (S2 vs S3), «transportadores ABC» fue la vía de enriquecimiento más significativa, seguida de «purina m». En la comparación de pares S3 vs. S4, ‘microbiana m. en ambientes diversos’ fue la vía de enriquecimiento más significativa, seguida por ‘ carbapenem m.’. En la comparación de pares S4 vs. S5, un número relativamente grande de vías mostraron resultados similares (dos metabolitos y valores de p similares), pero es notable la aparición de ‘isoflavonoide b.’ y ‘flavonoide b.’ entre ellas (Fig. 5; Cifras Complementarias).

LB metabolómico de datos: enriquecimiento de la vía metabólica de KEGG por pares entre etapas. (A) Etapa 1 vs.etapa 2. (B) Etapa 2 vs.etapa 3. (C) Etapa 3 vs etapa 4. (D) Etapa 4 vs la etapa 5. El factor rico es la relación entre el número de metabolitos regulados significativamente en la vía y el número total de metabolitos anotados en esa vía (rango = 0 a 1,0). El tamaño del punto representa el número de metabolitos significativamente enriquecidos en la vía correspondiente, y el color del punto representa el valor P (ambas leyendas se muestran en la figura).
Por lo que se refiere a los metabolitos individuales, en la comparación S1 vs.S2, la lista de metabolitos regulados al alza en el S1 estaba coronada por Trehalosa, Glactinol y ácido L-málico (todos ≈20 log2FC). El ácido oleico, el ácido 2-oxoadípico y el ácido esteárico fueron los metabolitos con mayor regulación ascendente en S2 (todos ≈15 a 18 log2FC). La lista de metabolitos con mayor regulación al alza en S2 en comparación con S3 fue coronada por dihidroxiacetona, LysoPC(18:1(9Z)) y Adenina (todos ≈16 a 17,5 log2FC). La lista de metabolitos con mayor regulación al alza en el S3 (en comparación con el S2) estuvo coronada por Trehalosa, Galactinol y ácido L-málico (todos ≈19 a 20 log2FC). En la comparación S3 vs. S4, la lista de metabolitos con mayor regulación al alza en el S3 fue coronada por ácido L-málico, DL-Arginina y ácido oleico (todos ≈18 a 19 log2FC), mientras que la lista de metabolitos con mayor regulación al alza en el S4 fue coronada por D-Manosa, N-Acetilactosamina y LisoPC(18:1(9Z)) en LB (todos ≈16 log2FC). En el S4 vs En comparación con S5, la lista de metabolitos con mayor regulación al alza en el S4 fue coronada por Trehalosa, ácido palmítico, N-acetilactosamina (todos ≈17 a 18 log2FC) en LB. La lista de metabolitos con mayor regulación al alza en el S5 fue coronada por L-Norleucina, ácido antranílico (Vitamina L1) y DL-Arginina (todos ≈16 a 18 log2FC) (Conjunto de Datos suplementarios S3).
Lycium ruthenicum (LR): análisis de pares entre etapas
Transcriptoma de LR
El análisis de mapas de calor de DEG en LR muestra que conjuntos de genes casi completamente diferentes estaban altamente regulados en las primeras etapas de desarrollo (1 + 2) y en la fruta madura (etapa 5), con un aparente reinicio transcriptómico después de la segunda etapa (Fig. 3B). El análisis de la relación de la muestra indica que las muestras podrían dividirse en dos clados (etapas 1 + 2 y 3 + 4 + 5) , con el último clado subdividido en dos clados: etapas 3 + 4 y etapa 5. El análisis de clasificación funcional intraespecífica de KEGG de estos DEG identificó 35 vías de manera significativa (P < 0,05) reguladas de manera diferente entre diferentes etapas de desarrollo (Conjunto de datos suplementarios S2). La «fotosíntesis», el » ácido linoleico m.», el » alcaloide de isoquinolina b.», el «flavonoide b.» y el «almidón y sacarosa m» (Fig. 3B). El mayor número de Grados fueron identificados en la mayoría de las comparaciones por pares de la etapa 1 (1 vs 2 fue una excepción), y en la etapa 2 vs 5 comparación (todos >5,000 Grados; Tabla 2). Con mucho, el número más pequeño se identificó en la comparación S3 vs. S4 (39). En la comparación de pares S1 vs S2, «amino azúcar y nucleótido azúcar m.» fue la vía regulada de forma más diferenciada, seguida por «ácido linoleico m.» (Fig. 6). En la comparación de pares S2 vs S3, las vías reguladas de forma más significativa y diferenciada fueron ‘alcaloide de isoquinolina b.’, ‘tirosina m.’, ‘fenilpropanoide b’y’ flavonoide b.’. En ambas comparaciones por pares, el mayor número de DEG (>15 y >35, respectivamente) se identificó en la «m de almidón y sacarosa». En el S3 vs Comparación S4, ‘ácido (alfa)linoleico m.’ fue la vía regulada más significativa, pero el número de genes fue mucho menor. En la comparación S4 vs. S5 ,el ‘ fenilpropanoide b ‘(también el mayor número de DEGs) y el’ ácido linoleico m ‘ fueron las vías reguladas más significativamente.
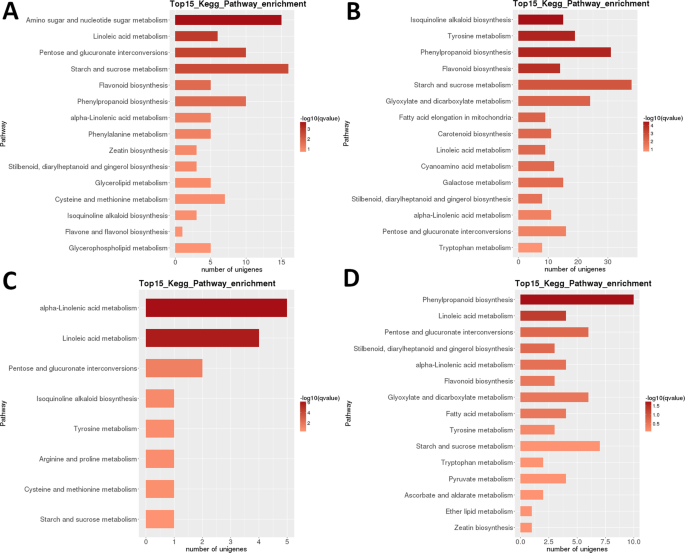
Transcriptoma LR: enriquecimiento de la vía metabólica de KEGG entre etapas por pares. (A) Etapa 1 vs.etapa 2. (B) Etapa 2 vs.etapa 3. (C) Etapa 3 vs etapa 4. (D) Etapa 4 vs la etapa 5. Se muestran las 15 vías más enriquecidas (o todas si el total es <15). El color de la barra representa la magnitud del valor q (un valor p ajustado por FDR, gráfico de colores que se muestra en la figura).
Metaboloma LR
El número de metabolitos regulados diferencialmente en comparaciones por pares de etapas de desarrollo en LR varió de 66 (etapas 3 frente a 4) a 133 (etapas 2 frente a 5) (Conjunto de datos suplementario S3). En comparaciones sucesivas de estadios, el mayor número se observó entre el 1º y el 2º estadio (117). Estos se asignaron a un gran número de vías; con el mayor número de metabolitos asignados a «b de metabolitos secundarios», seguido de «digestión y absorción de proteínas», «b. de aminoácidos» y «flavonoide b». En la comparación S2 vs. S3 ,los transportadores ABC fueron la vía de enriquecimiento más significativa, seguida de la purina m. En la comparación de pares S3 vs. S4, ‘microbiana m. en ambientes diversos’ fue la vía de enriquecimiento más significativa, seguida por ‘ carbapenem m.’. En el S4 vs Comparación de pares S5, un número relativamente grande de vías mostraron resultados similares (2 metabolitos y valores de p similares), pero es notable la aparición de ‘isoflavonoide b.’ y ‘flavonoide b.’ entre ellos (Fig. 7; Cifras Complementarias).

Datos metabolómicos de LR: enriquecimiento de la vía metabólica de KEGG entre etapas por pares. (A) Etapa 1 vs.etapa 2. (B) Etapa 2 vs.etapa 3. (C) Etapa 3 vs etapa 4. (D) Etapa 4 vs la etapa 5. El factor rico es la relación entre el número de metabolitos regulados significativamente en la vía y el número total de metabolitos anotados en esa vía (rango = 0 a 1,0). El tamaño del punto representa el número de metabolitos significativamente enriquecidos en la vía correspondiente, y el color del punto representa el valor P (ambas leyendas se muestran en la figura).
Por lo que se refiere a los metabolitos individuales (conjunto de datos suplementarios S3), en el S1 vs. En comparación con S2, la lista de metabolitos con regulación ascendente en el S1 estuvo coronada por Trehalosa, Galactinol y ácido L-málico (≈19-21 log2FC), mientras que el ácido oleico, el ácido 2-Oxoadípico y el ácido esteárico fueron los metabolitos con mayor regulación ascendente en el S2 (≈15-18 log2FC). En la comparación S2 vs. S3, la dihidroxiacetona, el sulfato de Indoxilo y la N-Acetilactosamina fueron los metabolitos con mayor regulación al alza en el S2 (≈17-19, 5 log2FC), y la Trehalosa, el Galactinol y el ácido L-málico (≈19-21 log2FC) en el S3. En el S3 vs Comparación S4, el ácido L-málico, DL-Arginina y ácido oleico se regularon al alza en el S3 (≈16-19 log2FC), y la 1,7-Dimetilxantina, D-Manosa y N-Acetilactosamina (≈15-17 log2FC) en el S4. En la comparación S4 vs. S5, el mononucleótido de flavina, la Trehalosa y el ácido isoferúlico se regularon al alza en el S4 (≈18-20 log2FC), mientras que los metabolitos PG(16:0/18:1(9Z)), D-Prolina y DL-Arginina se regularon al alza (todos ≈16 a 18 log2FC).
Análisis comparativo interespecífico de DEGs en diferentes etapas de desarrollo
DEGs totales durante el desarrollo de la fruta
Comparación interespecífica de etapas pareadas (LR1 vs. LB1, LR2 vs. LB2, etc.) muestra que 928 DEG fueron compartidos por los cinco pares (Fig. 8A). El mayor número de DEG se identificó en el estadio 3 (3989), y el más bajo en el estadio 4 (2825) (Fig. 8B); mientras que los números más altos de DEG únicos de un par se observaron en los estadios 3 (574), 1 y 5 (ambos 554), y los más bajos en el estadio 4 (126) (Fig. 8A). Los números de DEG regulados al alza y a la baja fueron relativamente similares en cada una de las comparaciones de etapas por pares; por ejemplo, en la etapa 5, 1668 DEG se regularon al alza y 1670 DEG se regularon a la baja en LR en comparación con LB (Fig. 8B). Sin embargo, en las otras cuatro etapas, el número de genes regulados al alza fue ligeramente superior (de 93 a 189 grados).

Genes expresados diferencialmente (DEGs) entre frutos de L. barbarum (LB) y L. ruthenicum (LR). (A) Comparaciones detalladas por etapas (1 a 5) (LB vs.LR). B) El número de grados reglados al alza (rojo) y reglados a la baja (verde) en LR en comparación con LB en cinco etapas de desarrollo estudiadas.
Las vías del transcriptoma
El análisis de mapas de calor de los DEG indica que los frutos de dos especies exhiben perfiles de expresión génica muy diferentes durante todas las etapas de desarrollo, pero las réplicas biológicas exhibieron perfiles muy similares, lo que indica una cantidad limitada de variabilidad individual en cada etapa de desarrollo (Figuras suplementarias: Fig. S9). El análisis comparativo del enriquecimiento de las vías de KEGG muestra que solo algunas vías fueron consistentemente altamente enriquecidas (en términos de regulación génica) en LR en comparación con LB a lo largo de las cinco etapas de desarrollo (Fig. 9). En particular, la transducción de señales de hormonas vegetales (2ª más alta en S1, 8ª más alta en S2, la más alta en S3, S4 y S5) y la interacción planta-patógeno (la más alta en S1, la 3ª más alta en S2, la 2ª más alta en S3, la 4ª más alta en S4 y la 15ª más alta en S5) se regularon relativamente al alza en todas las etapas. La biosíntesis de fenilpropanoides (no en el top 15 en S1, la más alta en S2, la 3ª más alta en S3, la 7ª más alta en S4, la 11ª más alta en S5), la ubiquinona y otras biosíntesis de terpenoides y quinonas (no en el top 15 en S1, la 6ª más alta en S2, la 7ª más alta en S3, la 2ª más alta en S4, la 6ª más alta en S5) también se regularon relativamente al alza en todas las etapas, excepto en la primera. La vía de biosíntesis de flavonoides no fue altamente enriquecida en las etapas tempranas (no en el top 15 en S1, 14º más alto en S2), y altamente enriquecida en las etapas tardías (3º a 4º más alto durante las etapas 3 a 5). el metabolismo del ácido (alfa-)linoleico fue altamente enriquecido en las etapas medias (9º más alto en S1, 2º más alto en S2, 5º y 6º más alto en S3, 8º más alto en S4, no en el top 15 en S5).

Análisis comparativo del enriquecimiento de la vía metabólica de KEGG. Las 15 vías principales enriquecidas en L. ruthenicum en comparación con L. barbarum se muestran a la izquierda (rojo), y las enriquecidas en L. barbarum en comparación con L. ruthenicum a la derecha (verde). Las etapas de desarrollo (1-5) se indican en la figura. el valor q es un valor p ajustado por FDR.
Entre las vías reguladas a la baja en L. ruthenicum en comparación con L. barbarum (Fig. 9) se observaron cambios notables entre las primeras etapas (1 y 2), cuando el metabolismo del ácido cianoamino y la biosíntesis de carotenoides se regularon más a la baja, y las etapas tardías (4 y 5), cuando las interacciones de SNARE en el transporte vesicular, el metabolismo del nicotinato y la nicotinamida, y el metabolismo de la porfirina y la clorofila se regularon constantemente relativamente a la baja.
Genes individuales del transcriptoma
Entre los genes más expresados diferencialmente, algunos eran específicos de la etapa de desarrollo (es decir, altamente regulados diferencialmente solo en etapas tempranas o tardías del desarrollo), pero algunos estaban consistentemente altamente regulados diferencialmente a lo largo de las cinco etapas estudiadas (Conjunto de datos suplementarios S4). Varios genes relacionados con la inmunidad muy regulados en LR en comparación con LB en las primeras etapas de desarrollo se encuentran entre los ejemplos del patrón de expresión específico de la etapa de desarrollo: quitinasa fue el 2º mayor alza GRADOS en S1 (13.43 veces), el más alto en S2 (13.89 veces), pero en las etapas posteriores no fue un GR. Del mismo modo, el receptor EIX 1/2 también se reguló muy al alza en las primeras etapas, el más alto en S1 (13,70) y el 2º más alto en S2 (10,90), pero tampoco se identificó como DEG en etapas posteriores. Algunos genes relacionados con el crecimiento también exhibieron un patrón de expresión similar: la fosfoglicerato quinasa (PGK) estuvo entre los pocos genes con mayor regulación al alza en las tres primeras etapas (13.14, 12.87 y 12).77, respectivamente), pero tampoco se identificó como DEG en etapas posteriores. La subunidad 7/8 del complejo CCR4-NOT transcription (CNOT7/8) también mostró un patrón de expresión muy similar: altamente regulado en las tres primeras etapas, y no un grado en las etapas 4 y 5. Varios genes asociados a la biosíntesis de flavonoides y fenilpropanoides exhibieron un patrón de expresión específico de la etapa de desarrollo invertida, con expresión relativamente baja en las etapas iniciales y muy alta en las etapas posteriores. Los ejemplos son: dihidroflavonol 4-reductasa bifuncional / flavanona 4-reductasa (DFR), que se reguló ligeramente al alza en LR en S1 (2.25), no un GRADO en S2, altamente regulado en S3 (7,79), y el 3er gen con mayor regulación en S4 (14,25) y S5 (16,03). Un paralogue de este gen mostraron un casi idéntico patrón: ligeramente al alza en S1 (2.44), no es un GRADOS en la S2, altamente regulada en el S3 (7.40), 6-mayor alza GRADOS en S4 (13.26) y 5º en S5 (14.59). De manera similar, el flavonoide 3′,5′-hidroxilasa (F3’5’H) no fue un grado en las dos primeras etapas, altamente regulado en S3 (6.69), el 5º gen con regulación al alza más alto en S4 (13.42) y el 4º más alto en S5 (15.05). Los flavonoides O-metiltransferasa (OMT) no era un GRADOS en la S1, pero en el S2 ya mostraron un nivel medio-alto de la regulación positiva de nivel (4.32), por el S3 ya era el tercer mayor alza DEG (13.30), y fue la mayor alza del gen en el S4 (18.73) y S5 (18.10). La leucoantocianidina dioxigenasa (LDOX; biosíntesis de antocianinas) no fue un grado en S1 y S2, seguido de una regulación ascendente de alto a muy alto en etapas posteriores (5,63, 9,44, 11,56, respectivamente). Dos análogos de la chalcona sintasa (CHS y CHS2; biosíntesis de flavonoides) tampoco estaban altamente regulados en S1 y S2 (CHS2: no un DEG, CHS: -1.14 en S1, no es un GRADOS en S2), pero en S3–S5 ambos genes mostraron una medio-alto de la regulación positiva (CHS2: 5.32, 7.84, 6.00; y CHS: 4.67, 7.01, 6.82; respectivamente). Seleccionamos estos genes para el análisis qPCR, y los resultados son altamente congruentes con los datos de ARN-seq (Resultados suplementarios; Conjunto de datos suplementarios S5). Finalmente, la citoquinina deshidrogenasa, un gen relacionado con la biosíntesis de la zeatina, también se reguló cada vez más durante las últimas tres etapas (2,6-5,7).
Sin embargo, algunos genes se expresaron de manera consistente y diferente a lo largo de los cinco estadios estudiados. Los ejemplos también incluyeron algunos genes relacionados con la inmunidad, como dos análogos de la glutatión S-transferasa, altamente regulados en LR en comparación con LB en todas las etapas: 9.38 y 8.58 (todos los valores presentados como cambios de pliegue en el orden respectivo) en S1, 6.30 y 6.34 en S2, el 2do y 7to grado más alto de regulación ascendente en S3 (14.08 y 12.70), el 2do y 4to grado más alto en S4 (15.71 y 14.16) y 2º y 6º más altos en S5 (16.48 y 14.40). La proteína de resistencia a enfermedades de las plantas RPM1 también fue altamente regulada en las cinco etapas (S1 = 13.15; S2 = 12.08; S3 = 13.11, S4 = 12.81; S5 = 13:94). Entre los genes consistentemente expresados de manera diferente a lo largo de todas las etapas de desarrollo también se encontraban algunos relacionados con el metabolismo de los aminoácidos, pero su patrón se invirtió en comparación con los ejemplos anteriores: exhibieron una alta regulación a la baja en LR en comparación con LB. Ejemplos son la acetil-COA aciltransferasa 1( AAT1; degradación de valina, leucina e isoleucina), con un perfil temporal de regulación descendente cada vez más alta, comenzando desde -7.0 en el S1 hasta <−10 veces en las últimas tres etapas. La prolina iminopeptidasa, asociada con el metabolismo de la arginina y la prolina, se reguló a la baja en LR en todas las etapas: S1 = -9,75, S2 = -10,89 (3º-más alto), S3 = -11,05 (4º-más alto), S4 = -10,01 y S5 = -11,98 (3º-más alto). Finalmente, la 5–metiltetrahidropteroiltriglutamato-homocisteína metiltransferasa (metE) se reguló constantemente extremadamente a la baja en LR en todas las etapas: 2a más alta en S1 (-11.76), la más alta en S2 (-11.74), 3a más alta en S3 (-11.43), la más alta en S4 (-12.36) y 2a más alta en S5 (-12.83). Dos genes asociados a la replicación y transcripción de ADN también se regularon a la baja en LR en todas las etapas: proteína nuclear de unión a GTP Ran (RAN; -10,0 a -12,0) y factor de replicación A1 (RFA1) (-8,0 a -12,0). Algunos genes relacionados con el crecimiento y el estrés también se regularon constantemente a la baja en LR: ribonucleoproteína nuclear heterogénea A1/A3 (hnRNP; -7 a -11) y proteína de choque térmico de 70 kDa 1/8 (HSPA1_8) S1 = -4,95, S2 = -8,88, S3 = -11,48 (2º-más alta), S4 = -9,36, S5 = -12,89 (la más alta). Curiosamente, un gen relacionado con la biosíntesis de fenilpropanoides, la shikimato hidroxicinamoiltransferasa (HCT), también fue consistentemente altamente regulado a la baja en LR: S1 = -6.82, S2 = -8.14, S3 = -11.71 (el más alto), S4 = -11.00 (3er-más alto), S5 = -11.91 (4to-más alto). Sin embargo, un regulador clave de la biosíntesis de antocianinas, el factor de transcripción MYB114, fue altamente regulado en LR durante las cinco etapas de desarrollo: 6.11, 4.69, 7.47, 9.05 y 8.95 (S1–S5 respectivamente).
Vías metabólicas
También se realizó un análisis comparativo interespecífico por etapas de las vías metabólicas (Fig. 10). En la primera etapa de desarrollo (S1), identificamos 39 metabolitos regulados diferencialmente. Entre las 20 vías principales con las que se asociaron estos metabolitos, varias de ellas se asociaron con aminoácidos, pero el número total de metabolitos por vía fue relativamente pequeño (1-2), y los valores de P no sugirieron un alto nivel de significación (Fig. 10-panel 1). En particular, el metabolismo (m) de la vitamina B6 y el m microbiano en diversos entornos mostraron valores de P comparativamente altos, factor de enriquecimiento (FE) de 1,0 y 2 metabolitos identificados. En el S2, identificamos 58 metabolitos regulados diferencialmente, asociados con solo cuatro vías: triptófano m, biosíntesis de fenilpropanoides (b), b de fenilpropanoides (estas son dos vías diferentes en la base de datos KEGG), y fenilalanina, tirosina y triptófano b (todos FE = 1.0, 2-3 metabolitos y p < 0.5; Fig. 10-panel 2). En el S3, identificamos 59 metabolitos regulados diferencialmente, asociados con 19 vías, la mayoría con la FE 1.0, pero valores de P relativamente no significativos (>0.5; Fig. 10-panel 3). Las vías con un número relativamente alto de metabolitos (n = 5) fueron: digestión y absorción de proteínas, b de metabolitos secundarios de plantas, b de antibióticos y b de aminoácidos. En el S4, identificamos 58 metabolitos regulados diferencialmente, asociados a un gran número de vías, la mayoría con la FE 1.0, y valores de significación comparativamente altos (principalmente P > 0.5; Fig. 10-panel 4). Las vías con un número relativamente alto de metabolitos (n ≥ 3) fueron: fenilpropanoide b, fenilalanina, tirosina y triptófano b, glucosinato b, b de alcaloides derivados de la vía del shikimato y 2-oxocarboxílico m. En la fruta madura (S5), identificamos 39 metabolitos regulados diferencialmente, asociados a un gran número de vías, pero en su mayoría con valores de P bajos y solo 1 metabolito por vía (Fig. 10-panel 5). Vías con más de 1 metabolito fueron: digestión y absorción de proteínas, fenilpropanoide b, absorción mineral, carbono central m en cáncer, b de metabolitos secundarios, b de fenilpropanoides y ARNt de aminoácidos b. El análisis de componentes principales (PCA) de todos los datos (2 especies × 5 estadios × 5 réplicas biológicas) reveló una alta similitud entre réplicas biológicas (agrupamiento), y corroboró una notable variabilidad entre diferentes estadios de maduración de frutos para ambas especies (Fig. 10-panel 6).

Análisis comparativo del enriquecimiento de la vía metabólica de KEGG. Las 15 vías principales enriquecidas en L. ruthenicum en comparación con L. barbarum se muestran a la izquierda (rojo), y las enriquecidas en L. barbarum en comparación con L. ruthenicum a la derecha (verde). Las etapas de desarrollo (1-5) se indican en la figura. el valor q es un valor p ajustado por FDR.
Metaboloma – metabolitos individuales
La lista (Conjunto de datos suplementarios S6) de los metabolitos regulados de forma más diferenciada entre las dos especies, mostró cierta variación entre las cinco etapas de desarrollo del fruto. Curiosamente, la fructosa 1-fosfato fue el metabolito con mayor regulación al alza en LR, en comparación con LB, durante las cinco etapas: cambio log2 = 6,3, 7,6, 7,7, 8,1 y 6,5 (etapas 1 a 5 respectivamente). En cuanto a los metabolitos altamente regulados en LB, hubo más variación entre etapas: en el S1, las diferencias fueron bastante pequeñas, con el 9-Decenol como el metabolito con mayor regulación al alza (cambio log2 = 2,7; comparado con LR). Los análisis de S2 y S3 produjeron resultados muy congruentes, con fenol (3,7 y 3,2 respectivamente) como el metabolito con mayor regulación al alza. En el S4, el sulfato de indoxilo (4,7) fue el metabolito superior regulado al alza en LB. Se observó un cambio metabólico en la fruta madura (S5), donde la lista de metabolitos regulados al alza en LB estaba coronada por estearoilcarnitina (7.1), ácido metoxiacético (5.3), S-Metil-5′-tioadenosina (4.7), lisinopril (4.7), Adenosina 3′, fosfato cíclico 5 ‘ (cAMP) (4.7), etc. Otros metabolitos altamente regulados en LR (aparte de Fructosa 1-fosfato) fueron naringina (6.2), lauroil-CoA (4.8), L-Pneilalanina (4.6), etc.