La presentación clínica más común del hiperparatiroidismo primario (PHP) es la hipercalcemia asintomática, y el diagnóstico de PHP basado en la presencia de manifestaciones óseas como la osteítis fibrosa quística (OFC) es cada vez más infrecuente. La OFC se presenta en menos del 5% de los pacientes con PHP y sugiere una enfermedad más grave o de larga duración. La OFC se caracteriza por la aparición de dolor óseo asociado con el hallazgo de cambios radiográficos específicos, como aumento de la reabsorción ósea subperióstea en el tercio distal del radio y las falanges medias, adelgazamiento clavicular distal, cráneo con «sal y pimienta», quistes óseos y tumores marrones en huesos largos. Los tumores marrones son el resultado de la desmineralización ósea con activación de osteoclastos, microhemorragia y microfracturas, y se denominan así por su color típico, debido a los abundantes depósitos de hemosiderina. Histopatológicamente, existe una combinación de actividad osteoclástica y osteoblástica con formación de quistes y muchos macrófagos cargados de hemosiderina.1 El diagnóstico diferencial de tumores marrones incluye granuloma reparador de células gigantes y tumor de células gigantes (TCG) óseo.
Se reporta el caso de un paciente con PHP debido a un adenoma paratiroideo con tumores marrones que imitan un TCG metastásico.
Un niño de 47 años acudió por primera vez al servicio de cirugía ortopédica de un hospital de Castilla-La Mancha en mayo de 2008, quejándose de dolor en cadera y mano izquierda no asociado a traumatismo previo. El paciente relató antecedentes personales de dislipidemia, diabetes mellitus tipo 2, hipertensión arterial, obesidad grado I y cólico renal con cálculos de oxalato de calcio. Sus antecedentes familiares incluían a dos hijas que habían sido diagnosticadas y sometidas a cirugía de PSP debido a un adenoma. La radiografía simple de cadera y mano mostró una imagen quística polilobulada que infló y adelgazó el hueso cortical en el tercer hueso metacarpiano izquierdo y lesiones de la rama lítica supracetabular e ilioisquiopúbica izquierda. Una tomografía computarizada de la pelvis (noviembre de 2008) mostró lesiones grandes en ala ilíaca, isquion, rama púbica izquierda, ala sacra derecha y cuello femoral. Con base en estos hallazgos, en julio y septiembre de 2009 el paciente fue sometido a cirugía consistente en legrado y relleno con injerto autólogo y sustitutos óseos del tercer hueso metacarpiano izquierdo y de la lesión supracetabular izquierda. El laboratorio patológico reportó un TCG. Controles posteriores con TC y RM del tórax y la pelvis revelaron el agrandamiento de lesiones líticas polilobuladas y expansivas en la pelvis (Fig. 1), sacro, cuello femoral derecho y L5, con la aparición de una nueva lesión en cabeza femoral derecha y séptimo arco costal izquierdo (Fig. 2). Estos cambios se atribuyeron a la progresión del tumor metastásico. Debido al dolor persistente que impidió totalmente la deambulación, el paciente fue remitido a la unidad de tumores óseos del Hospital Universitario La Paz, donde una revisión de muestras patológicas llevó a la conclusión de que las lesiones óseas eran altamente sugestivas de granulomas reparadores de células gigantes e histológicamente indistinguibles de los tumores marrones. Por lo tanto, se descartó el hiperparatiroidismo. En noviembre de 2010, el paciente fue remitido al servicio de endocrinología, donde las pruebas de laboratorio adicionales arrojaron los siguientes resultados: calcio total 14 mg/dL, calcio corregido 13,2 mg/dL, calcio iónico 1,72 mmol/L, fosfato 1,9 mg/dL, magnesio 1,86 mg/dL, calcio urinario 968,60 mg/24h, creatinina 0,55 mg/dL, iPTH 535pg/ml, vitamina D 13ng/ml. La TC de cuerpo total mostró un nódulo de 1,5 cm de diámetro en la ubicación teórica de la glándula paratiroidea derecha, y una exploración de paratiroides con 20 MCI de TC 99-sestamibi reveló hallazgos consistentes con adenoma de paratiroides hiperfuncionante derecho. Además, se descartó la presencia de un feocromocitoma asociado con base en bioquímica y morfología. Se realizó paratiroidectomía derecha y se notificó una biopsia intraoperatoria como adenoma paratiroideo. El estudio histopatológico final confirmó la presencia de un adenoma paratiroideo de 4,5 g de peso y 2,2 cm×2 cm×1,9 cm de tamaño.
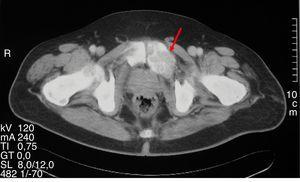
Figura 1.
Lesión lítica expansiva correspondiente a osteítis fibrosa quística en una tomografía computarizada pélvica.
(0.14 MB).
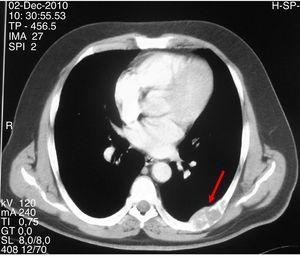
Figura 2.
Tomografía computarizada que muestra una lesión lítica expansiva con contorno lobulado y adelgazamiento cortical en el séptimo arco costal izquierdo.
(0.21 MB).
Después de la cirugía, el paciente experimentó hipocalcemia sintomática que requirió tratamiento con calcitriol y calcio, que se ha continuado hasta la fecha. Continúa siendo seguido en el departamento de endocrinología, reporta una mejoría sintomática significativa y es capaz de caminar con muletas. Un estudio genético no encontró mutaciones en el gen MEN-1.
El TCG es un tumor altamente vascularizado que se encuentra en las metáfisis o epífisis de los huesos largos o en la pelvis, el sacro o las vértebras.2 El aspecto radiográfico e histológico de los tumores marrones típicos de la OFC puede imitar de cerca un TCG, como ocurrió en nuestro paciente, y la diferenciación debe hacerse en función de los signos clínicos y los resultados de laboratorio (iPTH). Algunos de los autores3, 4 han notificado casos de OFC en los que inicialmente se sospechaba enfermedad ósea metastásica secundaria en función de los signos clínicos y las imágenes radiográficas. En nuestro paciente, sin embargo, el diagnóstico de tumor óseo primario metastásico se había basado en hallazgos histológicos, mientras que los antecedentes familiares, no notificados en los casos anteriores, eran consistentes con PHP.
Por otro lado,la deficiencia de vitamina D se detecta a menudo en pacientes con PHP e is3, 5 asociada a exacerbación de la presentación bioquímica y fenotípica de la enfermedad (niveles séricos más altos de PTH, adenomas paratiroideos grandes y mayor riesgo de fractura), lo que puede haber contribuido al florido cuadro clínico de nuestro paciente.
Se sabe que las formas familiares de hiperparatiroidismo son poco frecuentes (5%), y sus causas más frecuentes incluyen los síndromes de neoplasia endocrina múltiple (NEM) de tipo 1 y 2A, el síndrome de hiperparatiroidismo con tumor mandibular (HPT-JT) y el hiperparatiroidismo aislado familiar (FIHP).6 En HOMBRES 1, el hiperparatiroidismo es la presentación más temprana y frecuente (>90%), mientras que en HOMBRES 2A ocurre tarde y tiene una baja penetrancia. Aunque el estudio genético para HOMBRES 1 fue negativo en nuestro paciente, debe tenerse en cuenta que un resultado negativo falso puede ocurrir en hasta el 30% de los casos probados como resultado de patrones de mutación que involucran diferentes regiones génicas o mutaciones en genes aún desconocidos que afectan la transcripción o la acción de la menina.7 Esto, junto con la probable ocurrencia asincrónica de diferentes aspectos de MEN-1, hace necesaria la monitorización continua. El NEM 2A es poco probable en ausencia de compromiso neoplásico tiroideo o feocromocitoma. El diagnóstico diferencial también debe incluir HPT-JT debido a la escala de compromiso óseo y al gran tamaño del adenoma. El hallazgo final de un carcinoma de paratiroides habría apoyado este diagnóstico, debido a su frecuente aparición en HPT-JT.8 Sin embargo, la ausencia de lesiones fibro óseas mandibulares o maxilares y lesiones renales hizo que esto fuera poco probable. Por último, aunque la HP puede representar en algunos casos una variante de otros síndromes hiperparatiroideos, no se puede descartar la posibilidad de que mutaciones localizadas en loci aún no identificados distintos de los notificados en los HOMBRES 1 y 2 y en el HPT-JT puedan causar este síndrome.
El interés del caso relatado radica en el hecho de que ilustra la importancia de evaluar el metabolismo del fósforo y el calcio y la función paratiroidea en todos los pacientes con lesiones óseas, de sospechar un potencial PF si existen lesiones sugerentes y de buscar un probable componente genético subyacente a través de una historia familiar y personal detallada.