Nous avons collecté des fruits de L. barbarum et de L. ruthenicum à cinq stades de développement, des fruits jeunes (≈10 jours après la floraison) aux fruits mûrs (mûrs) (34-45 jours après la floraison), et a étudié leur transcriptome et leur métabolome.
- Assemblage RNA-seq de novo et annotation fonctionnelle des unigènes
- Lycium barbarum (LB): analyses interétages par paires
- Transcriptome LB
- Métabolome LB
- Lycium ruthène (LR): Analyses inter-étapes par paires
- Transcriptome LR
- Métabolome LR
- Analyse comparative interspécifique des degrés à différents stades de développement
- Degrés totaux pendant le développement du fruit
- Voies du transcriptome
- Gènes individuels du transcriptome
- Voies métabolomiques
- Métabolome – métabolites individuels
Assemblage RNA-seq de novo et annotation fonctionnelle des unigènes
Nous avons préparé un total de 30 banques d’ADNc à partir de fruits de L. barbarum et L. ruthenicum, avec trois répliques biologiques (trois fruits de trois arbres) à chaque point temporel: 2 espèces × 5 points temporels × 3 répliques biologiques. Les échantillons ont été étiquetés LB /LR (1-5) – (1-3), où LB est L. barbarum et LR est L. ruthenicum, 1-5 sont des stades de développement du fruit (S1–S5), et 1-3 sont des échantillons individuels (répliques biologiques); ainsi, par exemple, LB1-1 représente L. barbarum, 1er stade de développement échantillonné (S1), échantillon de fruit No 1 (sur trois). Nous avons généré plus de 1,72 milliard de lectures de fin de paire pour ces 30 bibliothèques d’ADNc, ce qui correspond à une moyenne de 57,2 millions de lectures par échantillon (Jeu de données supplémentaire S1). Une évaluation rigoureuse de la qualité et un filtrage des données ont donné un total de 801 766 lectures de haute qualité avec une longueur moyenne de 730 et N50 de 1 107 pb (tableau 1). Enfin, un total de 326 276 unigènes de longueur moyenne de 596 pb et N50 de 847 pb ont été obtenus à partir des transcriptions (tableau 1). Les coefficients de corrélation des données ARN-seq pour les 30 échantillons indiquent une très bonne cohérence des résultats entre les répliques biologiques (Fig. 2).

Heatmap of correlation coefficients for RNA-seq data for 30 samples of L. barbarum (LB) and L. ruthenicum (LR) fruits at five different developmental stages. Les échantillons sont étiquetés LB/R1-5_1-3, où LB est L. barbarum, LR est L. ruthenicum, 1-5 sont des stades de développement des fruits et 1-3 échantillons individuels. Les échantillons ont été regroupés par regroupement hiérarchique; les dendrogrammes au-dessus et à gauche de la carte thermique indiquent la parenté des échantillons.
Parmi les 326 276 unigènes interrogés dans des bases de données publiques, un total de 193 021 gènes et/ou protéines appariés (59,15 %) dans au moins une base de données, et 12 171 (3,73 %) ont été annotés dans toutes les bases de données. Le plus grand nombre d’unigènes (149 863, 45,93%) a été annoté dans la base de données NT, et le plus petit nombre (24 017; 7,36%) dans la base de données KOG.
Lycium barbarum (LB): analyses interétages par paires
Transcriptome LB
Les nombres les plus élevés de degrés ont été identifiés dans toutes les comparaisons par paires du 1er stade et dans la comparaison du 2e et du 5e stade (tous les > 10 000 degrés; Tableau 2). Les plus petits nombres ont été identifiés dans les comparaisons de la 3e contre la 4e et de la 4e contre la 5e étape (255-257). L’analyse par carte thermique des DEGs dans LB montre que des ensembles de gènes assez différents ont été fortement régulés à la hausse dans les premiers stades de développement (1 + 2) et dans les stades ultérieurs (3 à 5) (Fig. 3 BIS). L’analyse de la parenté des échantillons indique que les échantillons pourraient être divisés en deux clades (stades 1 + 2 et 3 + 4 + 5 ), ce dernier clade étant subdivisé en deux clades : les stades 3 +4 et le stade 5. L’analyse de classification fonctionnelle de KEGG intraspécifique de ces DEG a identifié 15 voies significativement régulées (P < 0,05) de manière différentielle entre les différents stades de développement (Jeu de données supplémentaire S2). La « transduction du signal des hormones végétales », la « biosynthèse des phénylpropanoïdes (b.) », le « métabolisme de l’acide linoléique (m.) », l’amidon et le saccharose m ont été particulièrement fortement régulés.’, et ‘zeatin b.’ (fig. 3 BIS).

Heatmaps and functional pathway analyses of differentially expressed genes (DEG) in Lycium barbarum (panel A) and L. ruthenicum (panel B) fruits. Les cartes thermiques ont été générées par une analyse hiérarchique des degrés (axe y) et des échantillons individuels (axe x), où les dendrogrammes au-dessus et à gauche de la carte thermique indiquent la parenté des échantillons. Les échantillons sont étiquetés LB/R_1–5_1-3, où l’acronyme de l’espèce (LB ou LR) est suivi du stade de développement du fruit (1-5) et du numéro d’échantillon (1-3). Les analyses intraspécifiques de la voie du KEGG des DEG dans les cinq stades de développement des deux espèces sont indiquées à droite des cartes thermiques. Seules les 15 meilleures voies enrichies sont répertoriées. la valeur q est une valeur p ajustée par le FDR.
Pour une analyse plus approfondie des données, nous nous sommes concentrés sur la comparaison des voies les plus réglementées de manière significative dans les stades de développement successifs. Dans la première comparaison par paires (S1 vs S2), « phénylpropanoïde b. » était la voie de régulation différentielle la plus élevée, suivie de « amidon et saccharose m. » (Fig. 4). Un très grand nombre de DEGs (> 100) ont été identifiés dans les deux voies. Un résultat similaire a été observé dans la comparaison paire suivante, S2 vs S3, mais malgré le nombre assez important de DEGs (> 80) ‘ d’amidon et de saccharose m.’a montré une valeur q légèrement inférieure. Dans la comparaison entre S3 et S4, la « fixation du carbone dans les organismes photosynthétiques » était la voie la plus réglementée, mais le nombre de gènes était beaucoup plus faible. Dans la dernière paire, S4 vs S5, ‘zéatine b.’, flavonoïde b.’, acide gras b.’ et ‘galactose m.’ étaient les voies les plus significativement régulées, mais aucune des voies ne présentait plus de deux degrés.

Transcriptome LB : enrichissement par paire de la voie métabolique du KEGG inter-étages. A) Stade 1 vs stade 2. B) Stade 2 c. étape 3. C) Stade 3 contre stade 4. D) Stade 4 contre stade 5. Top 15 (ou tous si total < 15) des voies significativement enrichies sont indiquées. La couleur de la barre représente l’amplitude de la valeur q (une valeur p ajustée par FDR). Le nuancier est illustré sur la figure.
Métabolome LB
Nous avons effectué des comparaisons par paires de stades de développement pour identifier l’enrichissement des métabolites entre toutes les paires de stades. Le nombre de métabolites à régulation différentielle dans les comparaisons par paires des stades de développement de LB variait de 66 (stades 3 vs 4) à 129 (stades 2 vs 5). Le plus grand nombre de comparaisons successives a été observé entre le 1er et le 2e stade (117). Ceux-ci ont été attribués à un grand nombre de voies; le plus grand nombre de métabolites étant attribué à « b. des métabolites secondaires » (également la valeur p la plus faible), suivi de « digestion et absorption des protéines » et de « b. des acides aminés » (Fig. 5; Chiffres supplémentaires). Dans la comparaison des paires d’étages successifs (S2 vs. S3), ‘transporteurs ABC’ était la voie la plus significativement enrichie, suivie de ‘purine m.’. Dans la comparaison des paires S3 et S4, « microbial m. in diverse environments » était la voie la plus enrichie de manière significative, suivie de « carbapenem m. ». Dans la comparaison de la paire S4 vs S5, un nombre relativement important de voies a montré des résultats similaires (deux métabolites et des valeurs de p similaires), mais l’apparition d' » isoflavonoïde b. » et de « flavonoïde b. » parmi eux est remarquable (Fig. 5; Chiffres supplémentaires).

Données métabolomiques LB: enrichissement par paire de la voie métabolique du KEGG interétage. A) Stade 1 vs stade 2. B) Stade 2 vs stade 3. C) Stade 3 contre stade 4. D) Stade 4 contre stade 5. Le facteur riche est le rapport entre le nombre de métabolites significativement régulés dans la voie et le nombre total de métabolites annotés dans cette voie (plage = 0 à 1,0). La taille du point représente le nombre de métabolites significativement enrichis dans la voie correspondante, et la couleur du point représente la valeur P (les deux légendes sont représentées sur la figure).
En ce qui concerne les métabolites individuels, dans la comparaison S1 vs S2, la liste des métabolites régulés à la hausse dans le S1 était complétée par le Tréhalose, le Glatinol et l’acide L-malique (tous ≈20 log2FC). L’acide oléique, l’acide 2-Oxoadipique et l’acide stéarique étaient les métabolites les plus fortement régulés dans S2 (tous ≈15 à 18 log2FC). La liste des métabolites les plus fortement régulés dans S2 par rapport à S3 était complétée par la Dihydroxyacétone, le LysoPC (18:1 (9Z)) et l’adénine (tous ≈16 à 17,5 log2FC). La liste des métabolites les plus fortement réglementés à la hausse dans le S3 (par rapport au S2) était complétée par le tréhalose, le Galactinol et l’acide L-malique (tous ≈19 à 20 log2FC). Dans la comparaison S3 vs S4, la liste des métabolites les plus fortement réglementés à la hausse dans le S3 était complétée par l’acide L-malique, la DL-Arginine et l’acide oléique (tous ≈18 à 19 log2FC), tandis que la liste des métabolites les plus fortement réglementés à la hausse dans le S4 était complétée par le D-Mannose, la N-Acétyllactosamine et le LysoPC (18: 1 (9Z)) dans LB (tous ≈16 log2FC). Dans le S4 vs. Comparaison S5, la liste des métabolites les plus fortement réglementés à la hausse dans le S4 était complétée par le tréhalose, l’acide palmitique, la N-Acétyllactosamine (tous ≈17 à 18 log2FC) en LB. La liste des métabolites les plus fortement réglementés dans le S5 était complétée par la L-Norleucine, l’acide anthranilique (Vitamine L1) et la DL-Arginine (tous ≈16 à 18 log2FC) (Ensemble de données supplémentaires S3).
Lycium ruthène (LR): Analyses inter-étapes par paires
Transcriptome LR
L’analyse par carte thermique des DEGs dans LR montre que des ensembles de gènes presque complètement différents ont été fortement régulés aux premiers stades de développement (1 + 2) et dans les fruits mûrs (stade 5), une réinitialisation transcriptomique apparente se produisant après la deuxième étape (Fig. 3B). L’analyse de la parenté des échantillons indique que les échantillons pourraient être divisés en deux clades (stades 1 + 2 et 3 + 4 + 5 ), ce dernier clade étant subdivisé en deux clades : les stades 3 +4 et le stade 5. L’analyse de la classification fonctionnelle de KEGG intraspécifique de ces DEG a identifié 35 voies de manière significative (P < 0,05) régulées de manière différentielle entre les différents stades de développement (Jeu de données supplémentaire S2). La « photosynthèse », « l’acide linoléique m. », « l’alcaloïde d’isoquinoléine b. », « le flavonoïde b. » et « l’amidon et le saccharose m » ont été particulièrement fortement régulés de manière différenciée (Fig. 3B). Les nombres les plus élevés de degrés ont été identifiés dans la plupart des comparaisons par paires de l’étape 1 (1 contre 2 était une exception) et dans la comparaison de l’étape 2 contre 5 (tous > 5 000 degrés; Tableau 2). De loin, le plus petit nombre a été identifié dans la comparaison S3 vs S4 (39). Dans la comparaison par paires S1 vs S2, « sucre aminé et sucre nucléotidique m. » était la voie la plus régulée de manière différentielle, suivie de « acide linoléique m. » (Fig. 6). Dans la comparaison par paires S2 vs S3, les voies régulées de manière différentielle les plus significatives étaient « l’alcaloïde b. d’isoquinoléine », « la tyrosine m. », « le phénylpropanoïde b » et « le flavonoïde b. ». Dans les deux comparaisons par paires, le plus grand nombre de degrés (> 15 et > 35 respectivement) a été identifié dans le « m amidon et saccharose ». Dans le S3 vs. Comparaison S4, « l’acide (alpha-) linoléique m. » était la voie la plus significativement régulée, mais le nombre de gènes était beaucoup plus faible. Dans la comparaison S4 vs S5, ‘phénylpropanoïde b’ (également le plus grand nombre de degrés) et ‘acide linoléique m.’ étaient les voies les plus significativement régulées.
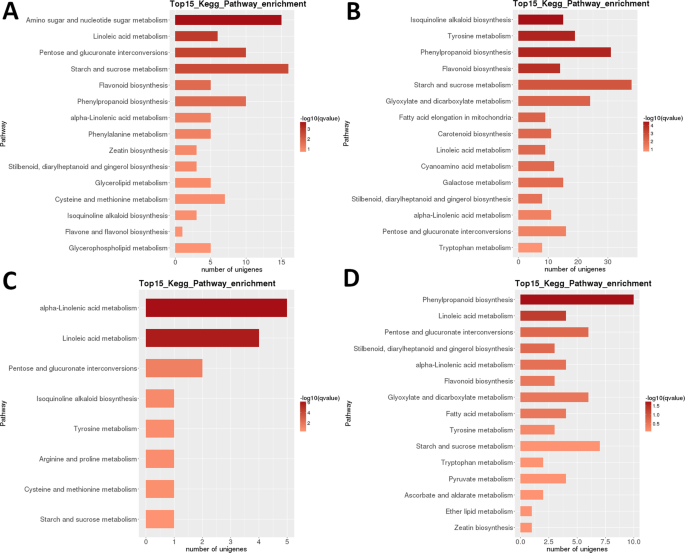
Transcriptome LR : enrichissement par paire de la voie métabolique du KEGG inter-étages. A) Stade 1 vs stade 2. B) Stade 2 vs stade 3. C) Stade 3 contre stade 4. D) Stade 4 contre stade 5. Top 15 (ou tous si total < 15) des voies significativement enrichies sont indiquées. La couleur de la barre représente l’amplitude de la valeur q (une valeur p ajustée par le FDR, nuancier illustré sur la figure).
Métabolome LR
Le nombre de métabolites à régulation différentielle dans les comparaisons par paires des stades de développement de LR variait de 66 (stades 3 vs 4) à 133 (stades 2 vs 5) (Jeu de données supplémentaire S3). Dans les comparaisons successives d’étapes, le plus grand nombre a été observé entre le 1er et le 2e stade (117). Ceux-ci ont été affectés à un grand nombre de voies; avec le plus grand nombre de métabolites affectés à « b des métabolites secondaires », suivi de « digestion et absorption des protéines », « b. des acides aminés » et « flavonoïde b ». Dans la comparaison entre S2 et S3, ‘transporteurs ABC’ était la voie la plus enrichie, suivie de ‘purine m’. Dans la comparaison des paires S3 et S4, « microbial m. in diverse environments » était la voie la plus enrichie de manière significative, suivie de « carbapenem m. ». Dans le S4 vs. Comparaison des paires S5, un nombre relativement important de voies a montré des résultats similaires (2 métabolites et des valeurs de p similaires), mais l’apparition d' »isoflavonoïde b. » et de « flavonoïde b. » parmi eux est remarquable (Fig. 7; Chiffres supplémentaires).

Données métabolomiques LR: enrichissement par paire de la voie métabolique du KEGG entre les étapes. A) Stade 1 vs stade 2. B) Stade 2 vs stade 3. C) Stade 3 contre stade 4. D) Stade 4 contre stade 5. Le facteur riche est le rapport entre le nombre de métabolites significativement régulés dans la voie et le nombre total de métabolites annotés dans cette voie (plage = 0 à 1,0). La taille du point représente le nombre de métabolites significativement enrichis dans la voie correspondante, et la couleur du point représente la valeur P (les deux légendes sont représentées sur la figure).
En ce qui concerne les métabolites individuels (jeu de données supplémentaire S3), dans le S1 vs. En comparaison avec S2, la liste des métabolites réglementés à la hausse dans le S1 était complétée par le Tréhalose, le Galactinol et l’acide L-Malique (≈19-21 log2FC), tandis que l’acide oléique, l’acide 2-Oxoadipique et l’acide stéarique étaient les métabolites les plus réglementés à la hausse dans le S2 (≈15-18 log2FC). Dans la comparaison S2 vs S3, la Dihydroxyacétone, le sulfate d’Indoxyle et la N-Acétyllactosamine étaient les métabolites les plus fortement régulés dans le S2 (≈17-19,5 log2FC), et le Tréhalose, le Galactinol et l’acide L-malique (≈19-21 log2FC) dans le S3. Dans le S3 vs. Comparaison avec le S4, l’acide L-malique, la DL-Arginine et l’acide oléique ont été régulés à la hausse dans le S3 (≈16-19 log2FC), et la 1,7-Diméthylxanthine, le D-Mannose et la N-Acétyllactosamine (≈15-17 log2FC) dans le S4. Dans la comparaison S4 vs S5, la flavine mononucléotide, le tréhalose et l’acide isoférulique étaient régulés à la hausse dans les métabolites S4 (≈18-20 log2FC), tandis que la PG (16: 0/18:1 (9Z)), la D-Proline et la DL-Arginine étaient les métabolites les plus fortement régulés (tous ≈16 à 18 log2FC).
Analyse comparative interspécifique des degrés à différents stades de développement
Degrés totaux pendant le développement du fruit
Comparaison interspécifique par deux stades (LR1 vs LB1, LR2 vs LB2, etc.) montre que 928 degrés ont été partagés par les cinq paires (Fig. 8 BIS). Le plus grand nombre de DEGs a été identifié à l’étape 3 (3989) et le plus faible à l’étape 4 (2825) (Fig. 8B); alors que les nombres les plus élevés de DEGRÉS uniques à une paire ont été observés aux étapes 3 (574), 1 et 5 (554 toutes deux), et les plus faibles à l’étape 4 (126) (Fig. 8 BIS). Les nombres de degrés régulés à la hausse et à la baisse étaient relativement similaires dans chacune des comparaisons d’étages par paires; par exemple, à l’étape 5, 1668 degrés ont été régulés à la hausse et 1670 degrés ont été régulés à la baisse en LR par rapport à LB (Fig. 8B). Cependant, dans les quatre autres stades, le nombre de gènes régulés à la hausse était légèrement plus élevé (93 à 189 degrés).

Gènes exprimés de manière différentielle (DEGs) entre les fruits de L. barbarum (LB) et L. ruthenicum (LR). (A) Comparaisons détaillées par étapes (1 à 5) (LB vs LR). (B) Le nombre de degrés régulés à la hausse (rouge) et à la baisse (vert) dans LR par rapport à LB dans cinq stades de développement étudiés.
Voies du transcriptome
L’analyse par carte thermique des DEGs indique que les fruits de deux espèces présentent des profils d’expression génique très différents à tous les stades de développement, mais que les répliques biologiques présentent des profils très similaires, indiquant une quantité limitée de variabilité individuelle à chaque stade de développement (Figures supplémentaires: Fig. S9). L’analyse comparative de l’enrichissement de la voie de KEGG montre que seules certaines voies étaient constamment fortement enrichies (en termes de régulation génétique) en LR par rapport à LB tout au long des cinq stades de développement (Fig. 9). Notamment, la transduction du signal des hormones végétales (2e plus élevée en S1, 8e plus élevée en S2, la plus élevée en S3, S4 et S5) et l’interaction plante-agent pathogène (la plus élevée en S1, la 3e plus élevée en S2, la 2e plus élevée en S3, la 4e plus élevée en S4 et la 15e plus élevée en S5) étaient relativement fortement régulées à tous les stades. La biosynthèse des phénylpropanoïdes (pas dans le top 15 en S1, la plus élevée en S2, la 3e plus élevée en S3, la 7e plus élevée en S4, la 11e plus élevée en S5), l’ubiquinone et d’autres biosynthèses terpénoïdes-quinones (pas dans le top 15 en S1, la 6e plus élevée en S2, la 7e plus élevée en S3, la 2e plus élevée en S4, la 6e plus élevée en S5) ont également été relativement fortement régulées à tous les stades, sauf le premier. La voie de biosynthèse des flavonoïdes n’a pas été très enrichie aux stades précoces (pas dans le top 15 en S1, 14e – le plus élevé en S2), et très enrichie aux stades tardifs (3e à 4e – le plus élevé au cours des stades 3 à 5). Le métabolisme de l’acide (alpha-) linoléique a été fortement enrichi aux stades intermédiaires (9e – le plus élevé en S1, 2e-le plus élevé en S2, 5e et 6e – le plus élevé en S3, 8e-le plus élevé en S4, pas dans le top 15 en S5).

Analyse comparative de l’enrichissement de la voie métabolique du KEGG. Les 15 premières voies enrichies en L. ruthenicum par rapport à L. barbarum sont indiquées à gauche (rouge), et celles enrichies en L. barbarum par rapport à L. ruthenicum à droite (vert). Les stades de développement (1-5) sont indiqués sur la figure. la valeur q est une valeur p ajustée par le FDR.
Parmi les voies régulées à la baisse chez L. ruthenicum par rapport à L. barbarum (Fig. 9) des changements notables ont été observés entre les stades précoces (1 et 2), lorsque le métabolisme des cyanoaminoacides et la biosynthèse des caroténoïdes étaient les plus fortement régulés, et les stades tardifs (4 et 5), lorsque les interactions de PIÈGE dans le transport vésiculaire, le métabolisme du nicotinate et du nicotinamide, et le métabolisme de la porphyrine et de la chlorophylle étaient constamment relativement fortement régulés.
Gènes individuels du transcriptome
Parmi les gènes les plus fortement exprimés de manière différentielle, certains étaient spécifiques à un stade de développement (c’est-à-dire très régulés de manière différentielle seulement au début ou à la fin du développement), mais certains étaient constamment très régulés de manière différentielle tout au long des cinq stades étudiés (Jeu de données supplémentaire S4). Plusieurs gènes liés à l’immunité très fortement régulés en LR par rapport à LB dans les premiers stades de développement sont parmi les exemples du modèle d’expression spécifique au stade de développement: la chitinase était le 2e degré le plus élevé régulé à la hausse dans S1 (13,43 fois), le plus élevé dans S2 (13,89 fois), mais dans les stades ultérieurs, ce n’était pas un DEGRÉ. De même, le récepteur EIX 1/2 a également été très fortement régulé à la hausse dans les premiers stades, le plus élevé dans S1 (13,70) et le 2e plus élevé dans S2 (10,90), mais il n’a pas non plus été identifié comme DEG dans les stades ultérieurs. Certains gènes liés à la croissance présentaient également un schéma d’expression similaire: la phosphoglycérate kinase (PGK) était parmi la poignée de gènes les plus fortement régulés aux trois premiers stades (13.14, 12.87 et 12.77 respectivement), mais il n’a pas non plus été identifié comme DEG à des stades ultérieurs. La sous-unité 7/8 du complexe de transcription CCR4-NOT (CNOT7 / 8) présentait également un schéma d’expression très similaire: très régulé dans les trois premières étapes, et non pas un DEG dans les étapes 4 et 5. Plusieurs gènes associés à la biosynthèse des flavonoïdes et des phénylpropanoïdes ont présenté un schéma d’expression spécifique au stade de développement inversé, avec une expression relativement faible aux stades précoces et très élevée aux stades ultérieurs. Exemples: dihydroflavonol 4-réductase bifonctionnelle / flavanone 4-réductase (DFR), qui a été légèrement régulée à la hausse en LR dans S1 (2.25), pas un DEG dans S2, très régulé à la hausse dans S3 (7,79) et 3e gène le plus régulé à la hausse dans S4 (14,25) et S5 (16,03). Un parallogue de ce gène présentait un schéma presque identique: légèrement régulé à la hausse en S1 (2,44), pas un DEG en S2, très régulé à la hausse en S3 (7,40), 6e plus haut DEG régulé à la hausse en S4 (13,26) et 5e en S5 (14,59). De même, la flavonoïde 3′, 5′-hydroxylase (F3’5’H) n’était pas un DEG dans les deux premiers stades, fortement régulée à la hausse dans S3 (6,69), 5e gène régulé à la hausse dans S4 (13,42) et 4e gène le plus élevé dans S5 (15,05). La flavonoïde O-méthyltransférase (OMT) n’était pas un DEG dans le S1, mais dans le S2, elle présentait déjà un niveau de régulation à la hausse moyen-élevé (4,32), par le S3, c’était déjà le troisième DEG à la régulation à la hausse le plus élevé (13,30), et c’était le gène à la régulation à la hausse le plus élevé dans S4 (18,73) et S5 (18,10). La leucoanthocyanidine dioxygénase (LDOX; biosynthèse des anthocyanes) n’était pas un DEG dans S1 et S2, suivie d’une régulation à la hausse élevée à très élevée dans les stades ultérieurs (5,63, 9,44, 11,56, respectivement). Deux paralogues de chalcone synthase (CHS et CHS2; biosynthèse des flavonoïdes) n’étaient pas non plus très réglementés dans S1 et S2 (CHS2: pas un DEGRÉ, CHS: -1,14 dans S1, pas un DEG dans S2), mais dans S3-S5, les deux gènes présentaient une régulation à la hausse moyenne-élevée à élevée (CHS2: 5,32, 7,84, 6,00; et CHS: 4,67, 7,01, 6,82; respectivement). Nous avons sélectionné ces gènes pour l’analyse qPCR, et les résultats sont très congruents avec les données ARN-seq (Résultats supplémentaires; Jeu de données supplémentaires S5). Enfin, la cytokinine déshydrogénase, un gène lié à la biosynthèse de la zéatine, a également été de plus en plus régulée à la hausse au cours des trois dernières étapes (2,6-5,7).
Cependant, certains gènes étaient systématiquement exprimés de manière différentielle dans les cinq stades étudiés. Des exemples comprenaient également certains gènes liés à l’immunité, tels que deux paralogues de la glutathion S-transférase, fortement régulés à la hausse en LR par rapport à LB à tous les stades: 9,38 et 8,58 (toutes les valeurs présentées sous forme de changements de plis dans l’ordre respectif) en S1, 6,30 et 6,34 en S2, les 2e et 7e degrés les plus élevés régulés à la hausse en S3 (14,08 et 12,70), les 2e et 4e plus élevés en S4 (15,71 et 14.16) et 2ème et 6ème plus haut en S5 (16.48 et 14.40). La protéine de résistance aux maladies des plantes RPM1 a également été fortement régulée à la hausse dans les cinq stades (S1 = 13,15; S2 = 12,08; S3 = 13,11, S4 = 12,81; S5 = 13:94). Parmi les gènes constamment exprimés de manière différentielle à tous les stades de développement, certains étaient également liés au métabolisme des acides aminés, mais leur schéma a été inversé par rapport aux exemples précédents: ils présentaient une forte régulation négative de LR par rapport à LB. Des exemples sont l’acétyl-CoA acyltransférase 1 (AAT1; dégradation de la valine, de la leucine et de l’isoleucine), avec un profil temporel de régulation descendante de plus en plus élevée, allant de -7,0 dans le S1 à <− 10 fois dans les trois derniers stades. L’iminopeptidase de la proline, associée au métabolisme de l’arginine et de la proline, a été fortement régulée en LR à tous les stades: S1 = -9,75, S2 = -10,89 (3ème plus élevé), S3 = -11,05 (4ème plus élevé), S4 = -10,01 et S5 = -11,98 (3ème plus élevé). Enfin, la 5–méthyltétrahydroptéroyltriglutamate-homocystéine méthyltransférase (metE) a toujours été extrêmement fortement régulée à la baisse dans la LR à tous les stades: 2e plus élevé en S1 (-11,76), le plus élevé en S2 (-11,74), 3e plus élevé en S3 (-11,43), le plus élevé en S4 (-12,36) et 2e plus élevé en S5 (-12,83). Deux gènes associés à la réplication de l’ADN et à la transcription ont également été fortement régulés en LR à tous les stades : la protéine nucléaire de liaison au GTP Ran (RAN; -10,0 à -12,0) et le facteur de réplication A1 (RFA1) (-8,0 à -12,0). Certains gènes liés à la croissance et au stress ont également été constamment fortement régulés à la baisse dans LR : ribonucléoprotéine nucléaire hétérogène A1/A3 (hnRNP; -7 à -11) et protéine 1/8 de choc thermique de 70 kDa (HSPA1_8) S1 = -4,95, S2 = -8,88, S3 = -11,48 (2e plus élevé), S4 = -9,36, S5 = -12,89 (le plus élevé). Curieusement, un gène lié à la biosynthèse des phénylpropanoïdes, la shikimate hydroxycinnamoyltransférase (HCT), a également été constamment fortement régulé à la baisse en LR: S1 = -6,82, S2 = -8,14, S3 = -11,71 (le plus élevé), S4 = -11,00 (le 3e plus élevé), S5 = -11,91 (le 4e plus élevé). Cependant, un régulateur clé de la biosynthèse des anthocyanes, le facteur de transcription MYB114, a été fortement régulé à la hausse dans LR au cours des cinq stades de développement: 6,11, 4,69, 7,47, 9,05 et 8,95 (S1–S5 respectivement).
Voies métabolomiques
Nous avons également effectué une analyse comparative interspécifique des voies métaboliques (Fig. 10). Au premier stade de développement (S1), nous avons identifié 39 métabolites à régulation différentielle. Parmi les 20 principales voies auxquelles ces métabolites étaient associés, plusieurs d’entre elles étaient associées à des acides aminés, mais le nombre total de métabolites par voie était relativement faible (1-2), et les valeurs P ne suggéraient pas un niveau élevé de signification (Fig. 10 – panneau 1). Notamment le métabolisme de la vitamine B6 (m) et le m microbien dans divers environnements présentaient tous deux des valeurs de P relativement élevées, un facteur d’enrichissement (EF) de 1,0 et 2 métabolites identifiés. Dans le S2, nous avons identifié 58 métabolites à régulation différentielle, associés à seulement quatre voies: le tryptophane m, la biosynthèse des phénylpropanoïdes (b), b des phénylpropanoïdes (il s’agit de deux voies différentes dans la base de données KEGG) et la phénylalanine, la tyrosine et le tryptophane b (tous EF = 1,0, 2-3 métabolites et p < 0,5; Fig. 10 – panneau 2). Dans le S3, nous avons identifié 59 métabolites régulés de manière différentielle, associés à 19 voies, la plupart avec l’EF 1.0, mais des valeurs de P relativement non significatives (> 0,5; Fig. 10 – panneau 3). Les voies présentant un nombre relativement élevé de métabolites (n = 5) étaient les suivantes: digestion et absorption des protéines, b des métabolites secondaires des plantes, b des antibiotiques et b des acides aminés. Dans le S4, nous avons identifié 58 métabolites régulés de manière différentielle, associés à un grand nombre de voies, principalement avec l’EF 1.0, et des valeurs de signification relativement élevées (principalement P > 0,5; Fig. 10 – panneau 4). Les voies avec un nombre relativement élevé de métabolites (n ≥ 3) étaient les suivantes: phénylpropanoïde b, phénylalanine, tyrosine et tryptophane b, glucosinate b, b d’alcaloïdes dérivés de la voie du shikimate et 2-oxocarboxylique m. Dans le fruit mûr (S5), nous avons identifié 39 métabolites régulés de manière différentielle, associés à un grand nombre de voies, mais principalement avec de faibles valeurs de P et seulement 1 métabolite par voie (Fig. 10 – panneau 5). Les voies avec plus de 1 métabolite étaient: digestion et absorption des protéines, phénylpropanoïde b, absorption des minéraux, carbone central m dans le cancer, b des métabolites secondaires, b des phénylpropanoïdes et aminoacie ARNt b. L’analyse en composantes principales (ACP) de toutes les données (2 espèces × 5 stades × 5 répétitions biologiques) a révélé une grande similitude entre les répliques biologiques (regroupement) et corroboré une variabilité notable entre les différents stades de maturation des fruits pour les deux espèces (Fig. 10 – panneau 6).

Analyse comparative de l’enrichissement de la voie métabolique du KEGG. Les 15 premières voies enrichies en L. ruthenicum par rapport à L. barbarum sont indiquées à gauche (rouge), et celles enrichies en L. barbarum par rapport à L. ruthenicum à droite (vert). Les stades de développement (1-5) sont indiqués sur la figure. la valeur q est une valeur p ajustée par le FDR.
Métabolome – métabolites individuels
La liste (Jeu de données supplémentaires S6) des métabolites les plus fortement régulés de manière différentielle entre les deux espèces présentait une certaine variation entre les cinq stades de développement des fruits. Curieusement, le fructose 1-phosphate était le métabolite le plus fortement régulé dans LR, comparé à LB, pendant les cinq stades: changement de pli log2 = 6,3, 7,6, 7,7, 8,1 et 6,5 (stades 1 à 5 respectivement). En ce qui concerne les métabolites fortement régulés en LB, il y avait plus de variations entre les stades: dans le S1, les différences étaient plutôt faibles, le 9-décénol étant le métabolite le plus fortement régulé à la hausse (changement de pli log2 = 2,7; par rapport au LR). Les analyses des S2 et S3 ont produit des résultats très congruents, le phénol (3,7 et 3,2 respectivement) étant le métabolite le plus fortement régulé à la hausse. Dans le S4, le sulfate d’indoxyle (4,7) était le principal métabolite régulé à la hausse en LB. Un changement métabolique a été observé dans le fruit mûr (S5), où la liste des métabolites régulés à la hausse en LB était complétée par la stéaroylcarnitine (7.1), l’acide méthoxyacétique (5.3), la S-Méthyl-5′-thioadénosine (4.7), le lisinopril (4.7), Adénosine 3′, phosphate 5′-cyclique (AMPc) (4.7), etc. Les autres métabolites fortement régulés en LR (à l’exception du fructose 1-phosphate) étaient la naringine (6,2), le lauroyl-CoA (4,8), la L-Phneylalanine (4,6), etc.