Dimethyl Sulfoxide Oxidations
Dimethyl sulfoxide-based oxidation of primary alcohols to aldehydes or secondary alcohols to ketones is a mild method that does not depend upon heavy metal oxidants. Here is a brief, chronological history of the procedure.
Index:
-
Swern Oxidation (trifluoroacetic anhydride)
-
Swern Oxidation (oxalyl chloride)
Kornblum Oxidation: (1959) Un tosylate primaire est chauffé à 150o pour provoquer le déplacement de SN2 par l’oxygène du diméthylsulfoxyde (DMSO) en présence de NaHCO3. Le mécanisme accepté à l’époque était une élimination E2 comme indiqué. Le sulfure de diméthyle (DMS) est le produit de réduction de la réaction. Ce travail a formé le tosylate à partir de l’iodure d’alkyle avec le tosylate d’argent. On peut supposer que le tosylate peut être préparé à partir de l’alcool qui est une voie plus directe. L’inconvénient de la procédure est la température élevée bien que le temps de contact ne soit que de quelques minutes. La question essentielle est de trouver une méthode pour générer le sel de sulfénate.

Modification de Barton: (1964) Barton et ses collègues ont pu générer des sels de sulfénate en traitant les chloroformiates d’alkyle avec du DMSO avec perte de CO2. L’addition de triéthylamine génère le produit d’oxydation. Cette procédure a amélioré les conditions difficiles de la procédure Kornblum. De plus, le chloroformiate est facilement disponible par traitement de l’alcool à oxyder avec un excès de phosgène.

Oxydation de Moffatt-Pfitzner: (1963) Cette variante d’une oxydation à base de diméthylsulfoxyde a été développée chez Syntex. L’activateur est préparé par protonation du dicyclohexylcarbodiimide (1, DCC) qui est attaqué par le diméthylsulfoxyde. L’intermédiaire 2 est à nouveau protoné pour faciliter l’addition de l’alcool oxygène sur l’atome de soufre. La dicyclohexyl urée stable 4 est formée avec le sel de sulfénate 3. Cette espèce subit un effondrement au composé carbonyle sous l’influence de l’anion dihydrogénophosphate. Bien que l’acide phosphorique soit un catalyseur acide efficace pour cette réaction, l’acide sulfurique, le chlorure d’hydrogène et l’acide trifluoroacétique ne le sont pas. Cependant, le trifluoroacétate de pyridinium est un catalyseur efficace. Il est essentiel que la base conjuguée de l’acide soit suffisamment basique pour effectuer la dernière étape de la réaction.

Mécanisme Torrsell: (1966) Le chimiste suédois Torrsell a démontré que le mécanisme de ces oxydations à base de DMSO ne procède pas par une β-élimination intermoléculaire. L’élimination est un processus intramoléculaire. Le sel sulfénate 5 a été préparé par la procédure Moffatt-Pfitzner (voir ci-dessus). Si le mécanisme de Kornblum s’appliquait, le sulfure de diméthyle obtenu serait dépourvu de deutérium (Expérience 1). Cependant, on a obtenu du sulfure de diméthyle -d1(7). Ce résultat est cohérent avec une base qui déprotonate le groupe méthyle attaché au soufre formant l’ylide 6. L’ylide se décompose par déprotonation intramoléculaire pour produire les produits observés.
Expérience 1:

Pour corroborer ce mécanisme, le deutérium a été incorporé dans le DMSO sous forme de DMSO-d6. En conséquence, la réaction a produit du sulfure de diméthyle -d5 (12) et de l’aldéhyde 11 non neutralisé (Expérience 2).
Expérience 2:

Oxydation Parikh-Doering: (1967) Cette oxydation utilise le complexe de trioxyde de soufre de pyridine (13) comme activateur du diméthylsulfoxyde. Le sulfate est le groupe partant dans le déplacement par l’alcool (primaire ou secondaire) dans l’intermédiaire 14. Le sulfénate 15 est décomposé par le mécanisme intramoléculaire (vide supra) pour donner un aldéhyde ou une cétone.

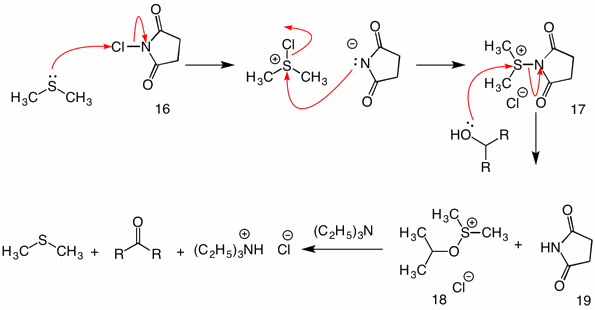
Oxydation de Corey-Kim: (1972) Dans ce procédé, le sulfure de diméthyle est activé (oxydé) avec le N-chlorosuccinimide (16) pour fournir le réactif 17. Dans la réaction de cette espèce avec un alcool, le groupe succinimidyle fonctionne comme un groupe partant. L’intermédiaire sulfénate usuel 18 s’effondre par le mécanisme intramoléculaire lors de l’addition de triéthylamine.
Oxydation de Swern : (1976) Cette oxydation précoce de Swern utilise l’anhydride trifluoroacétique (20) à -50oC pour activer le diméthylsulfoxyde. L’addition de l’alcool à l’intermédiaire 21 conduit au sulfénate 22 recherché. La cétone ou l’aldéhyde est produite de la manière habituelle avec la triéthylamine.

Oxydation de Swern: (1978) Cette procédure ultérieure de Swern est une méthode pratique pour la production du réactif 24 sans utiliser de sulfure de diméthyle et de chlore. Le diméthylsulfoxyde, qui est au même niveau d’oxydation que le sel 24, réagit avec le chlorure d’oxalyle (23) pour libérer le monoxyde de carbone, le dioxyde de carbone et le réactif 24. L’addition de l’alcool primaire ou secondaire suivie d’une déprotonation du sulfénate 25 avec de la triéthylamine conduit respectivement à l’aldéhyde ou à la cétone souhaités.
