La présentation clinique la plus courante de l’hyperparathyroïdie primaire (PHP) est l’hypercalcémie asymptomatique, et le diagnostic de PHP basé sur la présence de manifestations osseuses telles que l’ostéite fibrosa cystica (OFC) est de plus en plus rare. L’OFC survient chez moins de 5% des patients atteints de PHP et suggère une maladie plus grave ou de longue date. OFC se caractérise par l’apparition de douleurs osseuses associées à la découverte de modifications radiographiques spécifiques telles qu’une résorption osseuse sous-périostée accrue dans le tiers distal du radius et des phalanges moyennes, un amincissement claviculaire distal, un crâne « sel et poivre », des kystes osseux et des tumeurs brunes dans les os longs. Les tumeurs brunes résultent d’une déminéralisation osseuse avec activation des ostéoclastes, de microhémorragies et de microfractures, et sont ainsi nommées en raison de leur couleur typique, due à des dépôts abondants d’hémosidérine. Histopathologiquement, une combinaison d’activité ostéoclastique et ostéoblastique avec formation de kystes et de nombreux macrophages chargés d’hémosidérine existe.1 Le diagnostic différentiel des tumeurs brunes comprend le granulome réparateur à cellules géantes et la tumeur à cellules géantes (GCT) de l’os.
Le cas d’un patient atteint de PHP dû à un adénome parathyroïdien avec des tumeurs brunes imitant une GCT métastatique est rapporté.
Un homme de 47 ans s’est présenté pour la première fois au service de chirurgie orthopédique d’un hôpital de Castille-La Manche en mai 2008, se plaignant de douleurs à la hanche et à la main gauche non associées à un traumatisme antérieur. Le patient a signalé des antécédents personnels de dyslipidémie, de diabète sucré de type 2, d’hypertension artérielle, d’obésité de grade I et de coliques néphrétiques avec calculs d’oxalate de calcium. Ses antécédents familiaux comprenaient deux filles qui avaient été diagnostiquées et opérées pour PHP en raison d’un adénome. Une radiographie simple de la hanche et de la main a montré une image kystique polylobulée qui a gonflé et aminci l’os cortical du troisième os métacarpien gauche et des lésions lytiques du ramus ilioischiopubique supra-acétabulaire et gauche. Une tomodensitométrie du bassin (novembre 2008) a montré de grandes lésions au niveau de l’ala iliaque, de l’ischion, du ramus pubien gauche, de l’aile sacrée droite et du col fémoral. Sur la base de ces résultats, en juillet et septembre 2009, le patient a subi une intervention chirurgicale consistant en un curetage et un remplissage avec une greffe autologue et des substituts osseux du troisième métacarpien gauche et de la lésion supra-acétabulaire gauche. Le laboratoire de pathologie a signalé un GCT. Des contrôles ultérieurs avec tomodensitométrie et IRM de la poitrine et du bassin ont révélé l’élargissement de lésions lytiques polylobulées et expansives dans le bassin (Fig. 1), sacrum, col fémoral droit et L5, avec apparition d’une nouvelle lésion dans la tête fémorale droite et la septième arcade costale gauche (Fig. 2). Ces changements ont été attribués à la progression de la tumeur métastatique. En raison d’une douleur persistante qui empêchait totalement la déambulation, le patient a été dirigé vers l’unité des tumeurs osseuses de l’Hôpital Universitario La Paz, où un examen des échantillons pathologiques a conduit à la conclusion que les lésions osseuses étaient très évocatrices de granulomes réparateurs à cellules géantes et histologiquement indiscernables des tumeurs brunes. L’hyperparathyroïdie a donc été exclue. En novembre 2010, le patient a été référé au service d’endocrinologie, où des tests de laboratoire supplémentaires ont fourni les résultats suivants: calcium total 14 mg / dL, calcium corrigé 13,2 mg / dL, calcium ionique 1,72mmol / L, phosphate 1,9 mg / dL, magnésium 1,86 mg / dL, calcium urinaire 968,60 mg / 24h, créatinine 0,55 mg / dL, iPTH 535pg / mL, vitamine D 13ng / mL. La tomodensitométrie totale du corps a montré un nodule de 1,5 cm de diamètre à l’emplacement théorique de la glande parathyroïde droite, et une analyse parathyroïdienne avec 20 MCI de TC 99-sestamibi a révélé des résultats compatibles avec un adénome parathyroïdien hyperfonctionnel droit. De plus, la présence d’un phéochromocytome associé a été exclue sur la base de la biochimie et de la morphologie. Une parathyroïdectomie droite a été réalisée et une biopsie peropératoire a été signalée comme un adénome parathyroïdien. L’étude histopathologique finale a confirmé la présence d’un adénome parathyroïdien de 4,5 g de poids et de 2,2 cm× 2 cm× 1,9 cm de taille.
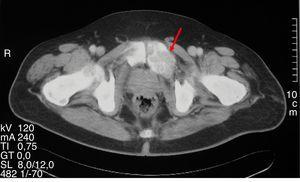
Figure 1.
Lésion lytique expansive correspondant à une ostéite fibrosa cystica lors d’une tomodensitométrie pelvienne.
(0.14 MB).
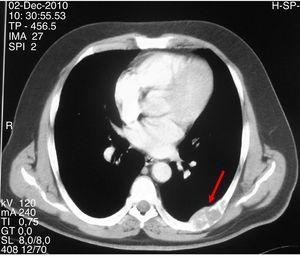
Figure 2.
Tomodensitométrie montrant une lésion lytique expansive avec contour lobulé et amincissement cortical dans la septième arcade costale gauche.
(0.21 MB).
Après la chirurgie, le patient a présenté une hypocalcémie symptomatique qui a nécessité un traitement avec du calcitriol et du calcium, qui a été poursuivi à ce jour. Il continue d’être suivi au service d’endocrinologie, signale une amélioration symptomatique significative et est capable de marcher avec des béquilles. Une étude génétique n’a trouvé aucune mutation dans le gène MEN-1.
La CTG est une tumeur hautement vascularisée qui se trouve dans les métaphyses ou les épiphyses des os longs ou dans le bassin, le sacrum ou les vertèbres.2 L’aspect radiographique et histologique des tumeurs brunes typiques de l’OFC peut imiter de près un GCT, comme cela s’est produit chez notre patient, et la différenciation doit être faite en fonction des signes cliniques et des résultats de laboratoire (iPTH). Certains des auteurs3,4 ont signalé des cas d’OFC dans lesquels une maladie osseuse métastatique secondaire était initialement suspectée sur la base de signes cliniques et d’images radiographiques. Chez notre patient, cependant, le diagnostic d’une tumeur osseuse primaire métastatique était basé sur des résultats histologiques, tandis que les antécédents familiaux, non rapportés dans les cas ci-dessus, étaient compatibles avec PHP.
D’autre part, une carence en vitamine D est souvent détectée chez les patients atteints de PHP et d’is3,5 associés à une exacerbation de la présentation biochimique et phénotypique de la maladie (taux sériques plus élevés de PTH, adénomes parathyroïdes importants et risque accru de fracture), ce qui peut avoir contribué au tableau clinique florissant de notre patient.
Les formes familiales d’hyperparathyroïdie sont connues pour être rares (5%) et leurs causes les plus fréquentes comprennent les syndromes de néoplasie endocrinienne multiple (MEN) de type 1 et 2A, le syndrome d’hyperparathyroïdie-tumeur de la mâchoire (HPT-JT) et l’hyperparathyroïdie isolée familiale (FIHP).6 Chez les HOMMES 1, l’hyperparathyroïdie est la présentation la plus précoce et la plus fréquente (> 90%), tandis que chez les HOMMES 2A, elle survient tardivement et présente une faible pénétrance. Bien que l’étude génétique pour les HOMMES 1 ait été négative chez notre patient, il convient de noter qu’un résultat faussement négatif peut survenir dans jusqu’à 30% des cas testés en raison de schémas de mutation impliquant différentes régions géniques ou de mutations dans des gènes encore inconnus affectant la transcription ou l’action de la ménine.7 Ceci, associé à l’occurrence asynchrone probable de différents aspects de MEN-1, rend une surveillance continue nécessaire. MEN 2A est peu probable en l’absence d’atteinte néoplasique thyroïdienne ou de phéochromocytome. Le diagnostic différentiel devrait également inclure HPT-JT en raison de l’ampleur de l’atteinte osseuse et de la grande taille de l’adénome. La découverte finale d’un carcinome parathyroïdien aurait étayé ce diagnostic, en raison de sa fréquence dans le HPT-JT.8 Cependant, l’absence de lésions fibro-osseuses mandibulaires ou maxillaires et de lésions rénales rendait cela peu probable. Enfin, bien que le FIHP puisse représenter dans certains cas une variante d’autres syndromes hyperparathyroïdes, la possibilité que des mutations localisées dans des loci encore non identifiés autres que ceux rapportés chez les HOMMES 1 et 2 et chez HPT-JT puissent provoquer ce syndrome ne peut être exclue.
L’intérêt du cas rapporté réside dans le fait qu’il illustre l’importance d’évaluer le métabolisme du phosphore et du calcium et la fonction parathyroïdienne chez tous les patients présentant des lésions osseuses, de suspecter un potentiel PHP si des lésions suggestives existent, et de rechercher une composante génétique sous-jacente probable à travers une histoire familiale et personnelle détaillée.