zebraliśmy owoce L. barbarum i L. ruthenicum na pięciu etapach rozwoju, od młodych owoców (≈10 dni po kwitnieniu) do dojrzałych (dojrzałych) owoców (34-45 dni po kwitnieniu) i zbadaliśmy ich transkryptom i metabolom.
- RNA-seq de novo assembly and functional annotation of unigenes
- Lycium barbarum (LB): pairwise interstage analyses
- lb Transcriptom
- lb Metabolome
- Lycium ruthenicum (LR): pairwise interstage analyses
- Transkryptom LR
- LR Metabolome
- międzyspecyficzna analiza porównawcza DEGs na różnych etapach rozwoju
- całkowite DEGs podczas rozwoju owoców
- Transcriptom-pathways
- Transkryptom – poszczególne geny
- metabolome – pathways
- Metabolome-individual metabolits
RNA-seq de novo assembly and functional annotation of unigenes
przygotowaliśmy łącznie 30 bibliotek cDNA z owoców L. barbarum i L. ruthenicum, z trzema replikatami biologicznymi (trzy owoce z trzech drzew) w każdym punkcie czasowym: 2 gatunki × 5 punktów czasowych × 3 replikaty biologiczne. Próbki oznaczono LB/LR(1-5)-(1-3), gdzie LB to L. barbarum, A LR to L. ruthenicum, 1-5 to stadia rozwojowe owoców (S1-S5–, A 1-3 to pojedyncze próbki (replikaty biologiczne); tak więc na przykład LB1-1 oznacza L. barbarum, pierwszy etap rozwoju próbki (S1), próbka owoców nr 1 (z trzech). Dla tych 30 bibliotek cDNA wygenerowaliśmy ponad 1,72 miliarda odczytów w parach, co odpowiada średnio 57,2 miliona odczytów na próbkę (dodatkowy zestaw danych S1). Rygorystyczna ocena jakości i filtrowanie danych przyniosły łącznie 801 766 wysokiej jakości odczytów o średniej długości 730 i N50 1107 bp (Tabela 1). Ostatecznie z transkryptów uzyskano łącznie 326 276 unigenów o średniej długości 596 bp i N50 847 bp (Tabela 1). Współczynniki korelacji dla danych RNA-seq dla 30 próbek wskazują na bardzo dobrą spójność wyników wśród replikatów biologicznych (Fig. 2).

Heatmapa współczynników korelacji dla danych RNA-seq dla 30 próbek owoców L. barbarum (LB) i L. ruthenicum (LR) w pięciu różnych stadiach rozwojowych. Próbki są oznaczone LB / R1-5_1-3, gdzie LB to L. barbarum, LR to L. ruthenicum, 1-5 to stadia rozwoju owoców, a 1-3 pojedyncze próbki. Próbki pogrupowano według hierarchicznego grupowania; dendrogramy powyżej i po lewej stronie mapy cieplnej wskazują na pokrewieństwo próbek.
spośród wszystkich 326 276 unigenów zapytanych o publiczne bazy danych, łącznie 193 021 (59,15%) dopasowanych genów i/lub białek w co najmniej jednej bazie danych, a 12 171 (3,73%) zostało przypisanych do wszystkich baz danych. Największą liczbę unigenów (149 863, 45,93%) zanotowano w bazie NT, A najniższą (24 017, 7,36%) w bazie KOG.
Lycium barbarum (LB): pairwise interstage analyses
lb Transcriptom
najwyższe liczby DEGs zidentyfikowano we wszystkich porównaniach parami pierwszego etapu oraz w porównaniu drugiego vs.piątego etapu (wszystkie > 10 000 DEGs; Tabela 2). Najmniejsze liczby zidentyfikowano w porównaniach stopnia 3. vs. 4. i 4. vs. 5. (255-257). Analiza Heatmap DEGs w LB pokazuje, że dość różne zestawy genów były wysoce regulowane we wczesnych stadiach rozwoju (1 + 2) i w późniejszych stadiach (3 do 5) (Fig. 3A). Analiza zależności próbki wskazuje, że próbki można podzielić na dwa klady (etapy 1 + 2 i 3 + 4 + 5), przy czym drugi Klad podzielił się na dwa klady: etap 3 + 4 i etap 5. Wewnątrzspecyficzna analiza klasyfikacji funkcjonalnej KEGG tych DEGs zidentyfikowała 15 szlaków znacząco (P < 0,05) różniących się między różnymi stadiami rozwoju (dodatkowy zestaw danych S2). Szczególnie silnie odmiennie regulowane były „transdukcja sygnału hormonu roślinnego”, ” biosynteza fenylopropanoidowa (b.)”, ” metabolizm kwasu linolowego (m.)”, ” skrobia i sacharoza m.”, oraz ” zeatin b. „(rys. 3A).

analizy Heatmaps i functional pathway of differentially expressed genes (DEGs) in Lycium barbarum (panel A) and L. ruthenicum (panel B) fruits. Mapy cieplne zostały wygenerowane przez hierarchiczną analizę DEGs (oś y) i pojedynczych próbek (oś x), gdzie dendrogramy powyżej i na lewo od mapy cieplnej wskazują na powiązanie próbek. Próbki są oznaczone LB / r_1–5_1-3, gdzie po akronimie gatunku (LB lub LR) następuje etap rozwoju owoców (1-5) i numer próbki (1-3). Wewnątrzspecyficzne analizy szlaku KEGG DEGs we wszystkich pięciu stadiach rozwojowych u dwóch gatunków są pokazane po prawej stronie map ciepła. Tylko top 15 wzbogacone ścieżki są wymienione. wartość q jest wartością P skorygowaną o FDR.
w celu bardziej dogłębnej analizy danych skupiliśmy się na porównaniu najbardziej znacząco regulowanych ścieżek w kolejnych etapach rozwoju. W pierwszym porównaniu parami (S1 vs S2) najbardziej zróżnicowanym szlakiem był ” fenylopropanoid b.”, a następnie ” skrobia i sacharoza m. „(Fig. 4). W obu szlakach zidentyfikowano bardzo dużą liczbę DEGs (>100). Podobny wynik zaobserwowano w poniższym porównaniu parami, S2 vs S3, ale pomimo dość dużej liczby DEGs (>80) ” skrobi i sacharozy m. wykazywał nieco niższą wartość Q. W porównaniu S3 i S4 „Wiązanie węgla w fotosyntetycznych organizmach” było najbardziej znaczącym regulowanym szlakiem, ale liczba genów była znacznie niższa. W ostatniej parze, S4 vs. S5, ” zeatyna b.”, flawonoid b.”, kwas tłuszczowy b. ” i „galaktoza m.” były najbardziej znacząco regulowanymi szlakami, ale żaden z szlaków nie wykazywał więcej niż dwóch DEGs.

transkryptom LB: parowe międzystopniowe wzbogacenie szlaku metabolicznego KEGG. A) Etap 1 vs.etap 2. B) Etap 2 vs. etap 3. (C) Etap 3 vs.Etap 4. (D) etap 4 vs.Etap 5. Top 15 (lub all if total <15) przedstawiono znacząco wzbogacone ścieżki. Kolor paska reprezentuje wielkość wartości q (skorygowaną przez FDR wartość p). Tabela kolorów jest pokazana na rysunku.
lb Metabolome
przeprowadziliśmy parami porównań fazy rozwojowej w celu identyfikacji wzbogacenia metabolitów między wszystkimi parami fazy. Liczba metabolitów o różnej regulacji w porównaniach parowych etapów rozwoju w LB wahała się od 66 (etapy 3 vs 4) do 129 (etapy 2 vs 5). Największą liczbę w kolejnych porównaniach etapowych zaobserwowano między I a II etapem (117). Zostały one przypisane do dużej liczby szlaków; z największą liczbą metabolitów przypisano ” b. metabolitów wtórnych „( również najniższą wartość p), a następnie „trawienie i wchłanianie białka” i „B. aminokwasów” (Fig. 5; Liczby Dodatkowe). W kolejnym etapie porównanie par (S2 vs. S3), „transportery ABC” były najistotniej wzbogaconym szlakiem, a następnie ” purine m.”. W porównaniu par S3 i S4, „microbial m. in diverse environments” był najbardziej znaczącym wzbogaconym szlakiem, a następnie ” carbapenem m.”. W porównaniu par S4 i S5 stosunkowo duża liczba szlaków wykazywała podobne wyniki (dwa metabolity i podobne wartości p), ale godne uwagi jest pojawienie się wśród nich „izoflawonoidu b.” i „flawonoidu b.” (Fig. 5; Liczby Dodatkowe).

LB metabolomic dane: parowe międzystopniowe wzbogacanie szlaku metabolicznego KEGG. A) Etap 1 vs.etap 2. B) Etap 2 vs etap 3. (C) Etap 3 vs.Etap 4. (D) etap 4 vs.Etap 5. Czynnikiem bogatym jest stosunek liczby znacząco regulowanych metabolitów w szlaku oraz całkowitej liczby metabolitów opisywanych w tym szlaku (zakres = 0 do 1,0). Wielkość punktu reprezentuje liczbę znacząco wzbogaconych metabolitów w odpowiedniej ścieżce, a kolor punktu reprezentuje wartość P (obie legendy pokazane na rysunku).
w odniesieniu do poszczególnych metabolitów, w porównaniu S1 i S2, listę metabolitów upregulowanych w S1 uzupełniono Trehalozą, Glactinolem i kwasem L-jabłkowym (wszystkie ≈20 log2FC). Kwas oleinowy, kwas 2-Oksoadypowy i kwas stearynowy były najbardziej wyregulowanymi metabolitami w S2 (wszystkie ≈15 do 18 log2FC). Listę metabolitów o największej podwyższonej wartości w S2 w porównaniu z S3 uzupełniono Dihydroksyacetonem, Lizopc (18: 1 (9Z)) i Adeniną (wszystkie ≈16 do 17,5 log2FC). Listę metabolitów o największej wartości w S3 (w porównaniu z S2) uzupełniono Trehalozą, Galaktinolem i kwasem L-jabłkowym (wszystkie ≈19 do 20 log2FC). W porównaniu S3 Z S4 listę najbardziej wyregulowanych metabolitów w S3 uzupełniono kwasem L-jabłkowym, DL-argininą i kwasem oleinowym (wszystkie ≈18 do 19 log2FC), podczas gdy listę najbardziej wyregulowanych metabolitów w S4 uzupełniono D-Mannozą, N-Acetylaktozaminą i Lizopc(18:1(9Z)) w LB (wszystkie ≈16 log2FC). W S4 vs. Porównanie S5, listę najbardziej wyregulowanych metabolitów w S4 uzupełniono Trehalozą, kwasem palmitynowym, N-Acetylaktozaminą (wszystkie ≈17 do 18 log2FC) w LB. Listę najbardziej wyregulowanych metabolitów w S5 uzupełniono o L-Norleucynę, kwas antranilowy (witaminę L1) i DL-argininę (wszystkie ≈16 do 18 log2FC) (dodatkowy zestaw danych S3).
Lycium ruthenicum (LR): pairwise interstage analyses
Transkryptom LR
analiza Heatmapy DEGs w LR pokazuje, że prawie zupełnie różne zestawy genów były wysoko regulowane we wczesnych stadiach rozwoju (1 + 2) i dojrzałych owocach (Etap 5), z pozornym resetem transkryptomicznym występującym po drugim etapie (Fig. 3b). Analiza zależności próbki wskazuje, że próbki można podzielić na dwa klady (etapy 1 + 2 i 3 + 4 + 5), przy czym drugi Klad podzielił się na dwa klady: etap 3 + 4 i etap 5. Wewnątrzspecyficzna analiza klasyfikacji funkcjonalnej KEGG tych DEGs zidentyfikowała 35 szlaków znacząco (P < 0,05) różniących się między różnymi stadiami rozwoju (dodatkowy zestaw danych S2). Szczególnie silnie odmiennie regulowane były „fotosynteza”, ” kwas linolowy m.”, ” alkaloid izochinolinowy b.”, „flawonoid b. „oraz” skrobia i sacharoza m ” (Fig. 3b). Najwyższe liczby DEGs zidentyfikowano w większości porównań par etapu 1 (wyjątek stanowił 1 vs. 2) oraz w porównaniu etapu 2 vs. 5 (Wszystkie >5000 DEGs; Tabela 2). Zdecydowanie najmniejszą liczbę zidentyfikowano w porównaniu S3 i S4 (39). W porównaniu parami S1 i S2, „amino sugar i nucleotide sugar m.” był najbardziej zróżnicowanym szlakiem, a następnie ” linoleic acid m. „(Fig. 6). W porównaniu parami S2 i S3 najbardziej znaczącymi różnicowymi szlakami były ” alkaloid izochinolinowy b.”, ” tyrozyna m.”, „fenylopropanoid b.” i ” flawonoid b.”. W obu porównaniach par największą liczbę DEGs (odpowiednio>15 i >35) stwierdzono w ” skrobi i sacharozie m.”. W S3 vs. W porównaniu z S4, „kwas (alfa-)linolowy m.” był najistotniej regulowanym szlakiem, ale liczba genów była znacznie niższa. W porównaniu S4 i S5, „fenylopropanoid b” (również największa liczba DEGs) i „kwas linolowy m.” były najbardziej istotnie regulowanymi szlakami.
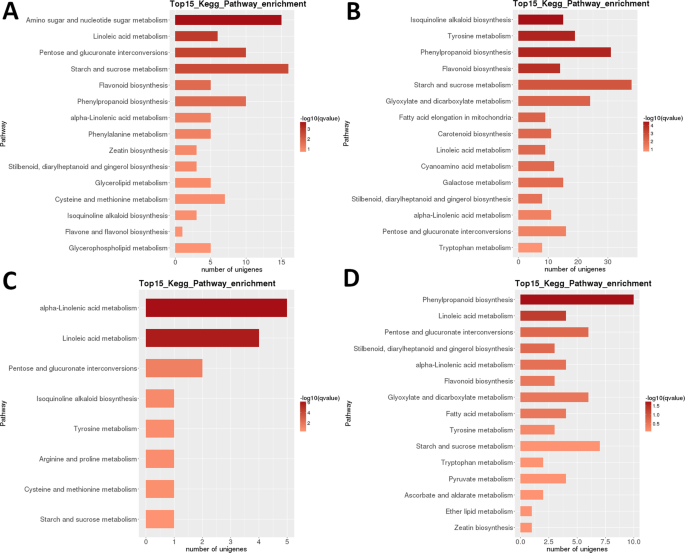
transkryptom LR: parowe międzystopniowe wzbogacenie szlaku metabolicznego KEGG. A) Etap 1 vs.etap 2. B) Etap 2 vs etap 3. (C) Etap 3 vs.Etap 4. (D) etap 4 vs.Etap 5. Top 15 (lub all if total <15) przedstawiono znacząco wzbogacone ścieżki. Kolor słupka reprezentuje wielkość wartości q (skorygowana przez FDR wartość p, wykres kolorów pokazany na rysunku).
LR Metabolome
liczba metabolitów regulowanych różnie w porównaniach parami etapów rozwoju w LR wahała się od 66 (etapy 3 vs 4) do 133 (etapy 2 vs 5) (dodatkowy zestaw danych S3). W kolejnych porównaniach etapowych największą liczbę obserwowano między I a II etapem (117). Zostały one przypisane do dużej liczby ścieżek; z największą liczbą metabolitów przypisanych do „B metabolitów wtórnych”, a następnie do „trawienia i wchłaniania białka”, „B. aminokwasów” i „flawonoidów b”. W porównaniu S2 vs. S3, „transportery ABC” był najbardziej znacząco wzbogacony szlak, a następnie „purine m”. W porównaniu par S3 i S4, „microbial m. in diverse environments” był najbardziej znaczącym wzbogaconym szlakiem, a następnie ” carbapenem m.”. W S4 vs. Porównanie par S5, stosunkowo duża liczba szlaków wykazywała podobne wyniki (2 metabolity i podobne wartości p), ale godne uwagi jest pojawienie się wśród nich „izoflawonoidu b.” i „flawonoidu b.” (Fig. 7; Dane Uzupełniające).

LR metabolomic dane: pairwise interstage KEGG metabolic pathway enrichment. A) Etap 1 vs.etap 2. B) Etap 2 vs etap 3. (C) Etap 3 vs.Etap 4. (D) etap 4 vs.Etap 5. Czynnikiem bogatym jest stosunek liczby znacząco regulowanych metabolitów w szlaku oraz całkowitej liczby metabolitów opisywanych w tym szlaku (zakres = 0 do 1,0). Wielkość punktu reprezentuje liczbę znacząco wzbogaconych metabolitów w odpowiedniej ścieżce, a kolor punktu reprezentuje wartość P (obie legendy pokazane na rysunku).
w odniesieniu do poszczególnych metabolitów (uzupełniający zestaw danych S3), w s1 vs. Porównanie S2, listę metabolitów upregulowanych w s1 uzupełniono o trehalozę, Galaktynol i kwas L-jabłkowy (≈19-21 log2FC), podczas gdy kwas oleinowy, kwas 2-Oksoadypowy i kwas stearynowy były najbardziej upregulowanymi metabolitami w S2 (≈15-18 log2FC). W porównaniu S2 i S3, dihydroksyaceton, siarczan Indoksylu i N-Acetylaktozamina były metabolitami o najwyższej wartości w S2 (≈17-19, 5 log2FC), a trehaloza, Galaktynol i kwas L-jabłkowy (≈19-21 log2FC) W S3. W S3 vs. Porównanie S4, kwas L-jabłkowy, DL-Arginina i kwas oleinowy były regulowane W S3 (≈16-19 log2FC), a 1,7-Dimetyloksantyna, D-Mannoza i N-Acetylaktozamina (≈15-17 log2FC) W S4. W porównaniu S4 i S5 mononukleotyd flawiny, trehaloza i kwas Izoferulowy ulegały podwyższonej regulacji w metabolitach S4 (≈18-20 log2FC), podczas gdy PG(16:0/18:1(9Z)), D-prolina i DL-arginina były najbardziej podwyższone (wszystkie ≈16-18 log2FC).
międzyspecyficzna analiza porównawcza DEGs na różnych etapach rozwoju
całkowite DEGs podczas rozwoju owoców
porównanie par międzyspecyficznych etapów (LR1 vs.LB1, LR2 vs. LB2 itp.) pokazuje, że 928 DEGs były wspólne dla wszystkich pięciu par (rys. 8a). Największą liczbę DEGs zidentyfikowano w etapie 3 (3989), a najniższą w etapie 4 (2825) (Fig. 8B); podczas gdy najwyższe liczby DEGs unikalne dla pary zaobserwowano w etapach 3 (574), 1 i 5 (oba 554), a najniższe w etapie 4 (126) (Fig. 8a). Liczby w górę iw dół regulowane DEGs były stosunkowo podobne w każdym z porównań par etapie; na przykład, w etapie 5, 1668 DEGs były regulowane i 1670 DEGs były regulowane w LR w porównaniu do LB (Fig. 8b). Jednak w pozostałych czterech stadiach liczba upregulowanych genów była nieznacznie wyższa (od 93 do 189 DEGs).

geny różnią się ekspresją (DEGs) między owocami L. barbarum (LB) i L. ruthenicum (LR). A) szczegółowe porównania stagewise (1 do 5) (LB vs. LR). (B) Liczba degS regulowanych w górę (czerwony) i w dół (zielony) w LR w porównaniu do LB w pięciu badanych stadiach rozwojowych.
Transcriptom-pathways
analiza Mapy termicznej DEGs wskazuje, że owoce dwóch gatunków wykazują bardzo różne profile ekspresji genów na wszystkich etapach rozwoju, ale replikaty biologiczne wykazują bardzo podobne profile, wskazując na ograniczoną zmienność indywidualną w każdym etapie rozwoju (dodatkowe figury: Fig. S9). Analiza porównawcza wzbogacania szlaku KEGG pokazuje, że tylko niektóre szlaki były konsekwentnie wysoce wzbogacone (pod względem regulacji genów) w LR w porównaniu do LB we wszystkich pięciu stadiach rozwoju (Fig. 9). W szczególności transdukcja sygnału hormonu roślinnego (2.-najwyższa w S1, 8.-najwyższa w S2, najwyższa w s3, S4 i S5) i interakcja roślina-patogen (najwyższa w S1, 3.-najwyższa w S2, 2.-najwyższa w S3, 4.-najwyższa w S4 i 15. – najwyższa w S5) były stosunkowo wysoko regulowane we wszystkich stadiach. Biosynteza fenylopropanoidów (Nie w top 15 W S1, najwyższa w S2, 3-najwyższa w S3, 7-najwyższa w S4, 11-najwyższa w S5), ubichinon i inne biosynteza terpenoidów-chinonów (Nie w top 15 W S1, 6-najwyższa w S2, 7-najwyższa w S3, 2-najwyższa w S4, 6-najwyższa w S5) były również stosunkowo wysoko regulowane we wszystkich etapach z wyjątkiem pierwszego. Szlak biosyntezy flawonoidów nie był wysoko wzbogacony we wczesnych stadiach (nie w top 15 W S1, 14-Najwyższy w s2), a wysoko wzbogacony w późnych stadiach (3-4-Najwyższy w stadiach 3-5). metabolizm kwasu (alfa-)linolowego był wysoce wzbogacony w fazie środkowej (9-Najwyższy w S1, 2-Najwyższy w S2, 5-i 6-Najwyższy w S3, 8-Najwyższy w S4, a nie w top 15 W S5).

Analiza porównawcza wzbogacenia szlaku metabolicznego KEGG. Pierwsze 15 ścieżek wzbogaconych w L. ruthenicum w porównaniu do L. Barbarum pokazano po lewej (czerwony), a te wzbogacone w L. Barbarum w porównaniu do L. ruthenicum po prawej (zielony). Etapy rozwoju (1-5) są wskazane na rysunku. wartość q jest wartością P skorygowaną o FDR.
wśród ścieżek w dół L. ruthenicum w porównaniu do L. barbarum (Fig. 9) zauważalne zmiany zaobserwowano między wczesnymi stadiami (1 i 2), gdy metabolizm kwasu cyjanoaminowego i biosynteza karotenoidów były najbardziej silnie regulowane w dół, a późnymi stadiami (4 i 5), gdy interakcje werbla w transporcie pęcherzykowym, metabolizm nikotynianu i nikotynamidu oraz metabolizm porfiryny i chlorofilu były konsekwentnie stosunkowo silnie regulowane w dół.
Transkryptom – poszczególne geny
wśród najbardziej różniących się genów, niektóre były specyficzne dla stadium rozwoju (tj. wysoce różniące się tylko we wczesnych lub późnych stadiach rozwoju), ale niektóre były konsekwentnie silnie różniące się we wszystkich pięciu badanych stadiach (uzupełniający zestaw danych S4). Kilka genów związanych z odpornością bardzo wysoko wyregulowanych w LR w porównaniu do LB we wczesnych stadiach rozwoju jest jednym z przykładów specyficznego dla etapu rozwoju wzorca ekspresji: chitynaza była drugim najwyższym stopniem w S1 (13,43-krotnie), Najwyższym w S2 (13,89-krotnie), ale w późniejszych stadiach nie była stopniem. Podobnie, receptor EIX 1/2 był również bardzo wysoko regulowany we wczesnych stadiach, Najwyższy w S1 (13,70) i drugi-najwyższy w S2 (10,90), ale nie został również zidentyfikowany jako DEG w późniejszych stadiach. Niektóre geny związane ze wzrostem również wykazywały podobny wzorzec ekspresji: kinaza fosfogliceratowa (PGK) była jedną z niewielu najbardziej regulowanych genów w pierwszych trzech etapach (13,14, 12,87 i 12.Odpowiednio 77), ale w późniejszych stadiach również nie został zidentyfikowany jako DEG. CCR4-nie transkrypcyjna złożona podjednostka 7/8 (CNOT7/8) również wykazywała bardzo podobny wzorzec ekspresji: silnie upregulowany w pierwszych trzech stadiach, a nie stopień w stadiach 4 i 5. Kilka genów związanych z biosyntezą flawonoidów i fenylopropanoidów wykazywało odwrócony wzór ekspresji specyficzny dla etapu rozwoju, ze stosunkowo niską ekspresją we wczesnych stadiach i bardzo wysoką w późniejszych stadiach. Przykładami są: dwufunkcyjna 4-reduktaza dihydroflawonolu/4-reduktaza flawononu (DFR), która została nieznacznie podwyższona w LR w S1 (2.25), bez stopnia w S2, silnie regulowany w S3 (7,79), a trzeci-najwyższy gen w S4 (14,25) i S5 (16,03). Paralogia tego genu wykazywała prawie identyczny wzór: nieznacznie podwyższony w S1 (2,44), Nie w S2, wysoce podwyższony w s3 (7,40), szósty najwyższy podwyższony W S4 (13,26) i piąty w S5 (14,59). Podobnie, flawonoid 3′, 5′-hydroksylaza (F3 ’ 5 ’ h) nie był stopniem w pierwszych dwóch etapach, silnie regulowany w S3 (6,69), piąty-najwyższy gen w S4 (13,42) i czwarty-najwyższy w S5 (15,05). O-metylotransferaza flawonoidowa (OMT) nie była stopniem w S1, ale w S2 wykazywała już średnio wysoki poziom regulacji w górę (4,32), przez S3 była już trzecim najwyższym stopniem regulacji w górę (13,30) i była najwyższym stopniem regulacji w S4 (18,73) i S5 (18,10). Dioksygenaza leukoantocyjanidynowa (ldox; biosynteza antocyjanów) nie była stopniem w S1 i S2, a następnie w późniejszych stadiach była regulowana od wysokiej do bardzo wysokiej (odpowiednio 5,63, 9,44, 11,56). Dwa paralogi syntazy chalkonowej (CHS i CHS2; biosynteza flawonoidów) również nie były silnie regulowane w S1 i S2 (CHS2: not A DEG, CHS: -1,14 w S1, Nie stopień w S2), ale w S3–S5 oba geny wykazywały średnio-wysoką DO WYSOKIEJ regulację (CHS2: 5.32, 7.84, 6.00; i CHS: 4.67, 7.01, 6.82; odpowiednio). Wybraliśmy te geny do analizy qPCR, a wyniki są bardzo zgodne z danymi RNA-seq (wyniki uzupełniające; dodatkowy zestaw danych S5). Wreszcie, dehydrogenaza cytokininy, gen związany z biosyntezą zeatyny, była również coraz bardziej regulowana w ostatnich trzech etapach (2,6–5,7).
jednak niektóre geny ulegały konsekwentnej ekspresji różnicowej we wszystkich pięciu badanych stadiach. Przykłady obejmowały również niektóre geny związane z odpornością, takie jak dwa paralogi s-transferazy glutationowej, silnie regulowane w LR w porównaniu do LB we wszystkich stadiach: 9,38 i 8,58 (wszystkie wartości przedstawione jako zmiany składane w odpowiedniej kolejności) w S1, 6,30 i 6,34 w S2, 2.i 7. najwyższe regulowane DEGs W S3 (14,08 i 12,70), 2. i 4. najwyższe W S4 (15,71 i 15,71). 14,16) oraz drugie i szóste najwyższe miejsce w S5 (16,48 i 14,40). Białko odpornościowe na choroby roślin RPM1 było również silnie regulowane we wszystkich pięciu stadiach (S1 = 13,15; S2 = 12,08; S3 = 13,11, S4 = 12,81; S5 = 13:94). Wśród genów konsekwentnie różniących się ekspresją na wszystkich etapach rozwoju były również niektóre związane z metabolizmem aminokwasów, ale ich wzór został odwrócony w porównaniu do poprzednich przykładów: wykazywały one wysoką regulację downregulacyjną w LR w porównaniu do LB. Przykładami są acylotransferaza acetylo-CoA 1 (aat1; degradacja waliny, leucyny i izoleucyny), o Profilu czasowym coraz bardziej wysokiej regulacji w dół, począwszy od -7,0 w S1 do <−10-krotnie w ostatnich trzech etapach. Iminopeptydaza prolinowa, związana z metabolizmem argininy i proliny, była silnie obniżona w LR we wszystkich stadiach: S1 = -9,75, S2 = -10,89 (trzeci najwyższy), S3 = -11,05 (czwarty najwyższy), S4 = -10,01 i S5 = -11,98 (trzeci najwyższy). Ostatecznie, 5–metylotetrahydropteroilotryglutaminian-metylotransferaza homocysteiny (metE) była konsekwentnie bardzo mocno obniżona w LR we wszystkich stadiach: 2. – najwyższa w S1 (-11,76), najwyższa w S2 (-11,74), 3.-najwyższa w S3 (-11,43), najwyższa w S4 (-12,36) i 2.-najwyższa w S5 (-12,83). Dwa geny związane z replikacją i transkrypcją DNA były również silnie obniżone w LR we wszystkich stadiach: białko jądrowe Ran wiążące GTP (RAN; -10,0 do -12,0) i czynnik replikacji A1 (RFA1) (-8,0 do -12,0). Niektóre geny związane ze wzrostem i stresem były również konsekwentnie silnie obniżane w LR: heterogeniczna rybonukleoproteina jądrowa A1/A3 (hnRNP; -7 do -11) i białko szoku cieplnego 70 kDa 1/8 (HSPA1_8) S1 = -4,95, S2 = -8,88, S3 = -11,48 (drugi najwyższy), S4 = -9,36, S5 = -12,89 (najwyższy). Co ciekawe, gen związany z biosyntezą fenylopropanoidową, shikimate hydroxycinnamoyltransferase (HCT), był również konsekwentnie silnie obniżany w LR: S1 = -6,82, S2 = -8,14, S3 = -11,71 (najwyższy), S4 = -11,00 (trzeci najwyższy), S5 = -11,91 (czwarty najwyższy). Jednak kluczowy regulator biosyntezy antocyjanów, czynnik transkrypcyjny MYB114, był silnie regulowany w LR we wszystkich pięciu etapach rozwoju: 6,11, 4,69, 7,47, 9,05 i 8,95 (odpowiednio S1–S5).
metabolome – pathways
przeprowadziliśmy również porównawczą międzygatunkową analizę etapową szlaków metabolicznych (Fig. 10). W pierwszym etapie rozwoju (S1) zidentyfikowaliśmy 39 metabolitów o różnej regulacji. Wśród 20 najważniejszych szlaków metabolitów, z którymi te metabolity były związane, kilka z nich było związanych z aminokwasami, ale całkowita liczba metabolitów na szlak była stosunkowo mała (1-2), A wartości P nie sugerowały wysokiego poziomu znaczenia (Fig. 10-panel 1). W szczególności metabolizm witaminy B6 (m) i drobnoustrojów m w różnych środowiskach wykazywał stosunkowo wysokie wartości P, współczynnik wzbogacania (EF) wynoszący 1,0 i 2 zidentyfikowane metabolity. W S2 zidentyfikowaliśmy 58 zróżnicowanych metabolitów, związanych tylko z czterema szlakami: tryptofanem m, biosyntezą fenylopropanoidów (b), B fenylopropanoidów (są to dwa różne szlaki w bazie danych KEGG) oraz fenyloalaniną, tyrozyną i tryptofanem b (wszystkie EF = 1,0, 2-3 metabolity i P < 0,5; Fig. 10-panel 2). W S3 zidentyfikowaliśmy 59 metabolitów o różnej regulacji, związanych z 19 szlakami, z których większość ma wartość EF 1,0, ale stosunkowo nieistotne wartości P (> 0,5; Fig. 10-panel 3). Szlaki o stosunkowo dużej liczbie metabolitów (n = 5) obejmowały: trawienie i wchłanianie białka, B metabolitów wtórnych roślin, B antybiotyków i B aminokwasów. W S4 zidentyfikowaliśmy 58 metabolitów o różnej regulacji, związanych z dużą liczbą szlaków, głównie z EF 1,0 i stosunkowo wysokimi wartościami istotności (głównie P > 0,5; Fig. 10-panel 4). Szlaki o stosunkowo dużej liczbie metabolitów (N ≥ 3) to: fenylopropanoid b, fenyloalanina, tyrozyna i tryptofan b, glukozynian b, alkaloidy B Pochodzące ze szlaku shikimate i 2-oksokarboksylowy m. w dojrzałych owocach (S5) zidentyfikowaliśmy 39 zróżnicowanych metabolitów, związanych z dużą liczbą szlaków, ale głównie z niskimi wartościami P i tylko 1 metabolitem na szlak (Fig. 10-panel 5). Szlaki z więcej niż 1 metabolitem były: trawienie i wchłanianie białka, fenylopropanoid b, wchłanianie minerałów, centralny węgiel m w raku, B wtórnych metabolitów, b fenylopropanoidów i aminoacy tRNA b. główna analiza składników (PCA) wszystkich danych (2 gatunki × 5 etapów × 5 replikatów biologicznych) ujawniła duże podobieństwo między replikatami biologicznymi (grupowanie) i potwierdziła znaczną zmienność między różnymi etapami dojrzewania owoców dla obu gatunków (Fig. 10-panel 6).

Analiza porównawcza wzbogacenia szlaku metabolicznego KEGG. Pierwsze 15 ścieżek wzbogaconych w L. ruthenicum w porównaniu do L. Barbarum pokazano po lewej (czerwony), a te wzbogacone w L. Barbarum w porównaniu do L. ruthenicum po prawej (zielony). Etapy rozwoju (1-5) są wskazane na rysunku. wartość q jest wartością P skorygowaną o FDR.
Metabolome-individual metabolits
the list (additional Dataset s6) of most highly differentially regulated metabolits between the two species, exhibited some variation among the five fruit development stages. Co ciekawe, 1-fosforan fruktozy był najbardziej regulowanym metabolitem w LR, w porównaniu do LB, podczas wszystkich pięciu etapów: Zmiana Log2 = 6,3, 7,6, 7,7, 8,1 i 6,5 (odpowiednio etapy 1 do 5). Jeśli chodzi o metabolity silnie regulowane w LB, odnotowano większą zmienność między etapami: w badaniu S1 różnice były raczej niewielkie, z 9-Decenolem jako metabolitem o najbardziej podwyższonej regulacji (Log2-krotna zmiana = 2,7; w porównaniu do LR). Analizy S2 i S3 dały bardzo przystające wyniki, z fenolem (odpowiednio 3,7 i 3,2) jako najbardziej wyregulowanym metabolitem. W S4 głównym metabolitem w LB był siarczan indoksylu (4.7). W dojrzałych owocach (S5) zaobserwowano przesunięcie metaboliczne, gdzie listę metabolitów upregulowanych w LB uzupełniały stearoilkarnityna (7.1), kwas Metoksyoctowy (5.3), s-metylo-5′-tioadenozyna (4.7), lizynopryl (4.7), adenozyna 3′, 5′-fosforan cykliczny (cAMP) (4.7) itp. Innymi metabolitami silnie regulowanymi w LR (oprócz 1-fosforanu fruktozy) były naringina (6.2), lauroylo-CoA (4.8), L-Phneylalanina (4.6) itd.