Die Illumina-Sequenzierungstechnologie verwendet Cluster-Generierung und Sequenzierung durch Synthese (SBS) -Chemie, um Millionen oder Milliarden von Clustern auf einer Flusszelle zu sequenzieren, abhängig von der Sequenzierungsplattform. Während der SBS-Chemie werden für jeden Cluster Basisaufrufe getätigt und für jeden Sequenzierungszyklus von der Echtzeitanalyse-Software (RTA) auf dem Instrument gespeichert. RTA speichert die Basisrufdaten in Form von einzelnen Basisrufdateien (oder BCL-Dateien). Wenn die Sequenzierung abgeschlossen ist, müssen die Basisaufrufe in den BCL-Dateien in Sequenzdaten konvertiert werden. Dieser Vorgang wird als BCL-zu-FASTQ-Konvertierung bezeichnet.
Eine FASTQ-Datei ist eine Textdatei, die die Sequenzdaten der Cluster enthält, die Filter auf einer Flusszelle passieren (weitere Informationen zu Clustern, die Filter passieren, finden Sie im Abschnitt „Zusätzliche Informationen“ dieses Bulletins). Wenn Samples gemultiplext wurden, ist der erste Schritt bei der Generierung von FASTQ-Dateien das Demultiplexen. Demultiplexing weist Cluster einer Stichprobe zu, basierend auf der/den Indexsequenz(en) des Clusters. Nach dem Demultiplexen werden die zusammengesetzten Sequenzen pro Sample in FASTQ-Dateien geschrieben. Wenn Proben nicht gemultiplext wurden, tritt der Demultiplexschritt nicht auf, und für jede Flusszellenbahn werden alle Cluster einer einzelnen Probe zugeordnet.
Für einen Single-Read-Lauf wird für jede Probe pro Durchflusszellenspur eine Read 1 (R1) FASTQ-Datei erstellt. Für einen gepaarten Endlauf wird für jede Probe für jede Spur eine R1- und eine Read 2 (R2) FASTQ-Datei erstellt. FASTQ-Dateien werden komprimiert und mit der Erweiterung * erstellt.schnelle Lieferung.gz.
Wie sieht eine FASTQ-Datei aus?
Für jeden Cluster, der den Filter durchläuft, wird eine einzelne Sequenz in die R1-FASTQ-Datei des entsprechenden Samples geschrieben, und für einen gepaarten Endlauf wird eine einzelne Sequenz auch in die R2-FASTQ-Datei des Samples geschrieben. Jeder Eintrag in einer FASTQ-Datei besteht aus 4 Zeilen:
- Eine Sequenzkennung mit Informationen über den Sequenzierungslauf und den Cluster. Der genaue Inhalt dieser Zeile hängt von der verwendeten BCL- zu FASTQ-Konvertierungssoftware ab.
- Die Sequenz (die Basis ruft; A, C, T, G und N).
- Ein Trennzeichen, das einfach ein Pluszeichen (+) ist.
- Die Basiswerte für die Anrufqualität. Diese sind Phred + 33 codiert und verwenden ASCII-Zeichen, um die numerischen Qualitätswerte darzustellen.
Hier ist ein Beispiel für einen einzelnen Eintrag in einer R1 FASTQ-Datei:
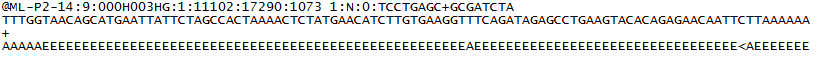
Weitere Informationen zum FASTQ-Format finden Sie hier.
Anzeigen einer FASTQ-Datei
FASTQ-Dateien können bis zu Millionen von Einträgen enthalten und mehrere Megabyte oder Gigabyte groß sein, was sie oft zu groß macht, um sie in einem normalen Texteditor zu öffnen. Im Allgemeinen ist es nicht erforderlich, FASTQ-Dateien anzuzeigen, da es sich um Zwischenausgabedateien handelt, die als Eingabe für Tools verwendet werden, die nachgelagerte Analysen durchführen, z. B. Ausrichtung auf eine Referenz oder De Novo-Assembly.
Wenn Sie eine FASTQ-Datei zur Fehlerbehebung oder aus Neugier anzeigen müssen, benötigen Sie entweder einen Texteditor, der sehr große Dateien verarbeiten kann, oder Zugriff auf ein Unix- oder Linux-System, auf dem große Dateien über die Befehlszeile angezeigt werden können.
Generieren von FASTQ-Dateien
Die Generierung von FASTQ-Dateien ist der erste Schritt für alle Analyse-Workflows, die von MiSeq Reporter auf dem MiSeq und Local Run Manager auf dem MiniSeq verwendet werden. Nach Abschluss der Analyse befinden sich die FASTQ-Dateien im <run folder>\Data\Intensities\BaseCalls auf dem MiSeq und im <output folder>\Alignment_#\<subfolder>\Fastq auf dem MiniSeq.
Für alle in BaseSpace Sequence Hub hochgeladenen Läufe erfolgt die Generierung von FASTQ-Dateien automatisch, nachdem der Lauf vollständig hochgeladen wurde, und die FASTQ-Dateien werden als Eingabe für die verschiedenen Analyse-Apps auf BaseSpace Sequence Hub verwendet. Auf BaseSpace Sequence Hub finden Sie Ihre FASTQ-Dateien in den Projekten, die Ihrem Lauf zugeordnet sind.
Mit der Konvertierungssoftware bcl2fastq können FASTQ-Dateien aus Daten generiert werden, die auf allen aktuellen Illumina-Sequenziersystemen generiert wurden.
Informationen zu den verschiedenen Einstellungen, die während der Generierung von FASTQ-Dateien angewendet werden können, finden Sie in den folgenden Software-Benutzerhandbüchern.
- MiSeq Reporter
Local Run Manager
bcl2fastq
Zusätzliche Informationen
- Eine Beschreibung und Anforderungen für Cluster, um Filter zu bestehen, finden Sie in Abschnitt 1.5.8 des Online-Schulungskurses MiSeq: Imaging and Base Calling.
- Weitere Informationen zu Basisanrufen auf NovaSeq-, NextSeq 500/550- und MiniSeq-Systemen finden Sie unter 2-Kanal-SBS-Technologie.
- Siehe Illumina Sequencing Technology für weitere Informationen über Basisanrufe auf MiSeq- und HiSeq-Systemen.