Illumina sequencing technology uses cluster generation and sequencing by synthesis (SBS) chemistry to sequence millions or billions of clusters on a flow cell, depending on the sequencing platform. Durante a química da SBS, para cada cluster, chamadas de base são feitas e armazenadas para cada ciclo de sequenciação pelo software de análise em tempo Real (RTA) no instrumento. A RTA armazena os dados de chamada de base na forma de arquivos de chamada de base individual (ou BCL). Quando a sequenciação completa, as chamadas de base nos arquivos BCL devem ser convertidas em dados de sequência. Este processo é chamado de conversão BCL para FASTQ.
um ficheiro FASTQ é um ficheiro de texto que contém os dados de sequência dos aglomerados que passam o filtro numa célula de fluxo (para mais informações sobre os aglomerados que passam o filtro, ver a secção “Informações adicionais” deste boletim). Se as amostras foram multiplexadas, o primeiro passo na geração de arquivos FASTQ é desmultiplexing. A desmultiplexação atribui aglomerados a uma amostra, com base na sequência de índice do aglomerado(s). Depois de desmultiplexar, as sequências montadas são escritas em arquivos FASTQ por amostra. Se as amostras não foram multiplexadas, o passo de desmultiplexagem não ocorre, e, para cada faixa de células de fluxo, todos os aglomerados são atribuídos a uma única amostra.
para uma execução de leitura única, um ficheiro FASTQ de leitura 1 (R1) é criado para cada Amostra por faixa de células de fluxo. Para uma execução emparelhada, um arquivo FASTQ R1 e um Read 2 (R2) é criado para cada amostra para cada faixa. Os arquivos FASTQ são comprimidos e criados com a extensão *.fastq.gz.
como é um ficheiro FASTQ?
para cada conjunto que passa o filtro, uma única sequência é escrita para o arquivo R1 FASTQ da amostra correspondente, e, para uma execução emparelhada, uma única sequência também é escrita para o arquivo R2 FASTQ da amostra. Cada entrada num ficheiro FASTQ consiste em 4 linhas:
- um identificador de sequência com informações sobre a sequência e o conjunto. O conteúdo exato desta linha varia com base no software de conversão BCL para FASTQ usado.
- a sequência (as chamadas de base; a, C, T, G E N).
- um separador, que é simplesmente um sinal de + ( + ).
- the base call quality scores. Estes são codificados Phred + 33, usando caracteres ASCII para representar as pontuações de qualidade numérica.
aqui está um exemplo de uma única entrada num ficheiro FASTQ R1:
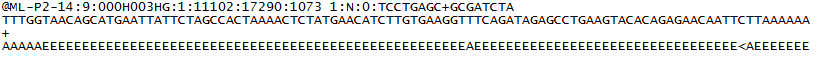
mais informações detalhadas sobre o formato FASTQ podem ser encontradas aqui.
como ver um ficheiro FASTQ
os ficheiros FASTQ podem conter até Milhões de Entradas e podem ter vários megabytes ou gigabytes de tamanho, O que frequentemente os torna demasiado grandes para serem abertos num editor de texto normal. Geralmente, não é necessário ver arquivos FASTQ, porque eles são Arquivos de saída intermediários usados como entrada para ferramentas que realizam análise a jusante, tais como alinhamento a uma referência ou montagem de novo.
se precisar de ver um ficheiro FASTQ para fins de resolução de problemas ou por curiosidade, irá precisar de um editor de texto que possa lidar com Ficheiros muito grandes, ou de acesso a um sistema Unix ou Linux onde ficheiros grandes possam ser visualizados através da linha de comandos.
como gerar arquivos FASTQ
a geração de arquivos FASTQ é o primeiro passo para todos os fluxos de análise usados por MiSeq Reporter no MiSeq e gerente de execução Local no MiniSeq. Quando a análise for concluída, o FASTQ arquivos estão localizados em <executar pasta>\Dados\Intensidades\BaseCalls no MiSeq e <pasta de saída>\Alignment_#\<subpasta>\Fastq no MiniSeq.
para todas as corridas carregadas para o Hub de sequência de BaseSpace, a geração de arquivos FASTQ ocorre automaticamente após a execução ser completamente carregada, e os arquivos FASTQ são usados como entrada para os vários aplicativos de análise no Hub de sequência de BaseSpace. No servidor de sequência BaseSpace, poderá encontrar os seus ficheiros FASTQ no(s) projecto (s) associado (s) à sua execução.
o software de conversão bcl2fastq pode ser usado para gerar arquivos FASTQ a partir de dados gerados em todos os sistemas de sequenciação ilumina atuais.
para informações sobre as diferentes configurações que podem ser aplicadas durante a geração de arquivos FASTQ, veja os guias de usuário do software abaixo.
- MiSeq Reporter
Local Run Manager
bcl2fastq
Additional information
- a description and requirements for clusters to pass filter can be found in section 1.5.8 of the MiSeq: Imaging and Base Calling online training course.
- See 2-Channel SBS Technology for more information about base calling on NovaSeq, NextSeq 500/550, and MiniSeq systems.
- See Illumina Sequencing Technology for more information about base calling on MiSeq and HiSeq systems.