La technologie de séquençage Illumina utilise la génération de grappes et le séquençage par chimie de synthèse (SBS) pour séquencer des millions ou des milliards de grappes sur une cellule à flux, selon la plate-forme de séquençage. Pendant la chimie SBS, pour chaque cluster, des appels de base sont effectués et stockés pour chaque cycle de séquençage par le logiciel d’Analyse en temps réel (RTA) sur l’instrument. RTA stocke les données d’appel de base sous la forme de fichiers d’appel de base individuels (ou BCL). Une fois le séquençage terminé, les appels de base dans les fichiers BCL doivent être convertis en données de séquence. Ce processus est appelé conversion BCL en FASTQ.
Un fichier FASTQ est un fichier texte qui contient les données de séquence des clusters qui passent le filtre sur une cellule de flux (pour plus d’informations sur les clusters qui passent le filtre, voir la section « informations supplémentaires » de ce bulletin). Si les échantillons ont été multiplexés, la première étape de la génération de fichiers FASTQ est le démultiplexage. Le démultiplexage attribue des clusters à un échantillon, en fonction de la ou des séquences d’index du cluster. Après le démultiplexage, les séquences assemblées sont écrites dans des fichiers FASTQ par échantillon. Si les échantillons n’ont pas été multiplexés, l’étape de démultiplexage ne se produit pas et, pour chaque voie de cellule d’écoulement, tous les clusters sont affectés à un seul échantillon.
Pour une exécution en lecture unique, un fichier FASTQ en lecture 1 (R1) est créé pour chaque échantillon par voie de cellules de flux. Pour une fin d’exécution appariée, un fichier FASTQ R1 et un fichier FASTQ Read 2 (R2) sont créés pour chaque échantillon pour chaque voie. Les fichiers FASTQ sont compressés et créés avec l’extension*.rapide.gz.
À quoi ressemble un fichier FASTQ?
Pour chaque cluster qui passe le filtre, une seule séquence est écrite dans le fichier FASTQ R1 de l’échantillon correspondant et, pour une exécution en bout apparié, une seule séquence est également écrite dans le fichier FASTQ R2 de l’échantillon. Chaque entrée dans un fichier FASTQ se compose de 4 lignes:
- Un identifiant de séquence contenant des informations sur l’exécution de séquençage et le cluster. Le contenu exact de cette ligne varie en fonction du logiciel de conversion BCL en FASTQ utilisé.
- La séquence (les appels de base; A, C, T, G et N).
- Un séparateur, qui est simplement un signe plus (+).
- Les scores de qualité d’appel de base. Ceux-ci sont codés Phred + 33, en utilisant des caractères ASCII pour représenter les scores de qualité numériques.
Voici un exemple d’une seule entrée dans un fichier FASTQ R1:
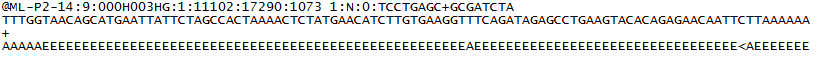
Des informations plus détaillées sur le format FASTQ peuvent être trouvées ici.
Comment afficher un fichier FASTQ
Les fichiers FASTQ peuvent contenir jusqu’à des millions d’entrées et peuvent avoir une taille de plusieurs mégaoctets ou gigaoctets, ce qui les rend souvent trop volumineux pour être ouverts dans un éditeur de texte normal. Généralement, il n’est pas nécessaire de visualiser les fichiers FASTQ, car ce sont des fichiers de sortie intermédiaires utilisés comme entrée pour des outils qui effectuent une analyse en aval, tels que l’alignement sur une référence ou un assemblage de novo.
Si vous avez besoin d’afficher un fichier FASTQ à des fins de dépannage ou par curiosité, vous aurez besoin soit d’un éditeur de texte capable de gérer des fichiers très volumineux, soit d’un accès à un système Unix ou Linux où les fichiers volumineux peuvent être visualisés via la ligne de commande.
Comment générer des fichiers FASTQ
La génération de fichiers FASTQ est la première étape pour tous les flux de travail d’analyse utilisés par MiSeq Reporter sur MiSeq et Local Run Manager sur MiniSeq. Une fois l’analyse terminée, les fichiers FASTQ se trouvent dans le dossier < run >\Data\Intensties\BaseCalls sur le MiSeq et le dossier de sortie < >\Alignment_#\< sous-dossier >\Fastq sur le MiniSeq.
Pour toutes les exécutions téléchargées sur BaseSpace Sequence Hub, la génération de fichiers FASTQ se produit automatiquement une fois l’exécution complètement téléchargée, et les fichiers FASTQ sont utilisés comme entrée pour les différentes applications d’analyse sur BaseSpace Sequence Hub. Sur BaseSpace Sequence Hub, vous pouvez trouver vos fichiers FASTQ dans le ou les projets associés à votre exécution.
Le logiciel de conversion bcl2fastq peut être utilisé pour générer des fichiers FASTQ à partir de données générées sur tous les systèmes de séquençage Illumina actuels.
Pour plus d’informations sur les différents paramètres pouvant être appliqués lors de la génération de fichiers FASTQ, consultez les guides d’utilisation du logiciel ci-dessous.
- MiSeq Reporter
Local Run Manager
bcl2fastq
Informations supplémentaires
- Une description et les exigences pour que les clusters passent le filtre se trouvent dans la section 1.5.8 du cours de formation en ligne MiSeq: Imagerie et appel de base.
- Voir Technologie SBS à 2 canaux pour plus d’informations sur les appels de base sur les systèmes NovaSeq, NextSeq 500/550 et MiniSeq.
- Voir Technologie de séquençage Illumina pour plus d’informations sur les appels de base sur les systèmes MiSeq et HiSeq.