Abbiamo raccolto i frutti della L. barbarum e L. ruthenicum a cinque fasi di sviluppo, dal frutto giovane (≈10 giorni post-fioritura) maturi (mature) frutta (34-45 giorni post-fioritura), e ha studiato il loro trascrittoma e metaboloma.
- RNA-seq de novo assemblaggio e annotazione funzionale di unigeni
- Lycium barbarum (LB): analisi interstadio a coppie
- Trascrittoma LB
- LB Metabolome
- Lycium ruthenicum (LR): analisi interstadiali a coppie
- Trascrittoma LR
- Metaboloma LR
- Analisi comparativa interspecifica di DEGs in diverse fasi di sviluppo
- DEGs totali durante lo sviluppo del frutto
- Transcriptome-pathways
- Trascrittoma – geni individuali
- Metabolome – pathways
- Metaboloma-singoli metaboliti
RNA-seq de novo assemblaggio e annotazione funzionale di unigeni
Abbiamo preparato un totale di 30 librerie di cDNA da frutti di L. barbarum e L. ruthenicum, con tre repliche biologiche (tre frutti da tre alberi) in ogni punto temporale: 2 specie × 5 punti temporali × 3 repliche biologiche. I campioni sono stati etichettati LB/LR (1-5)–(1-3), dove LB è L. barbarum e LR è L. ruthenicum, 1-5 sono stadi di sviluppo della frutta (S1-S5), e 1-3 sono campioni individuali (repliche biologiche); così per esempio LB1-1 rappresenta L. barbarum, 1st sampled developmental stage (S1), campione di frutta n.1 (su tre). Abbiamo generato oltre 1,72 miliardi di letture di coppia per queste 30 librerie cDNA, corrispondenti a una media di 57,2 milioni di letture per campione (Dataset supplementare S1). La rigorosa valutazione della qualità e il filtraggio dei dati hanno prodotto un totale di 801.766 letture di alta qualità con una lunghezza media di 730 e N50 di 1107 bp (Tabella 1). Infine un totale di 326.276 unigeni con la lunghezza media di 596 bp e N50 di 847 bp sono stati ottenuti dalle trascrizioni (Tabella 1). I coefficienti di correlazione per i dati RNA-seq per i 30 campioni indicano un’ottima consistenza dei risultati tra le repliche biologiche (Fig. 2).

Heatmap dei coefficienti di correlazione per i dati RNA-seq per 30 campioni di frutti L. barbarum (LB) e L. ruthenicum (LR) in cinque diversi stadi di sviluppo. I campioni sono etichettati LB / R1–5_1-3, dove LB è L. barbarum, LR è L. ruthenicum, 1-5 sono fasi di sviluppo della frutta e 1-3 singoli campioni. I campioni sono stati raggruppati per clustering gerarchico; i dendrogrammi sopra e a sinistra della mappa di calore indicano la parentela dei campioni.
Tra tutti i 326.276 unigenes interrogati su database pubblici, un totale di 193.021 (59,15%) ha abbinato geni e/o proteine in almeno un database e 12.171 (3,73%) sono stati annotati in tutti i database. Il maggior numero di unigenes (149.863, 45,93%) è stato annotato nel database NT e il numero più basso (24.017; 7,36%) nel database KOG.
Lycium barbarum (LB): analisi interstadio a coppie
Trascrittoma LB
Il numero più alto di DEG è stato identificato in tutti i confronti a coppie del 1 ° stadio e nel confronto tra il 2 ° e il 5 ° stadio (tutti > 10.000 DEG; Tabella 2). I numeri più piccoli sono stati identificati nei confronti della fase 3rd vs. 4th e 4th vs. 5th (255-257). L’analisi della mappa termica dei DEG in LB mostra che insiemi abbastanza diversi di geni erano altamente sovraregolati nelle prime fasi dello sviluppo (1 + 2) e nelle fasi successive (da 3 a 5) (Fig. 3 BIS). L’analisi della parentela del campione indica che i campioni potrebbero essere divisi in due cladi (fasi 1 + 2 e 3 + 4 + 5), con quest’ultimo clade ulteriormente suddiviso in due cladi: fasi 3 + 4 e fase 5. L’analisi di classificazione funzionale intraspecifica di KEGG di questi DEG ha identificato 15 percorsi in modo significativo (P < 0.05) differenzialmente regolati tra diversi stadi di sviluppo (Set di dati supplementari S2). Particolarmente fortemente differenzialmente regolati erano “trasduzione del segnale dell’ormone vegetale”, ” biosintesi fenilpropanoide (b.)”, ” metabolismo dell’acido linoleico (m.)”, ” amido e saccarosio m.”, e “zeatin b.” (Fig. 3 BIS).

Heatmap e analisi del percorso funzionale di geni espressi in modo differenziale (DEGs) nei frutti di Lycium barbarum (pannello A) e L. ruthenicum (pannello B). Le heatmap sono state generate da un’analisi gerarchica dei DEG (asse y) e dei singoli campioni (asse x), dove i dendrogrammi sopra e a sinistra della heatmap indicano la parentela dei campioni. I campioni sono etichettati LB/R_1–5_1–3, dove l’acronimo della specie (LB o LR) è seguito dallo stadio di sviluppo del frutto (1-5) e dal numero del campione (1-3). Le analisi intraspecifiche della via di KEGG di DEGs in tutte e cinque le fasi di sviluppo nelle due specie sono indicate alla destra delle heatmap. Sono elencati solo i primi 15 percorsi arricchiti. il valore q è un valore p regolato da FDR.
Per un’analisi più approfondita dei dati, ci siamo concentrati sul confronto dei percorsi più significativamente regolati nelle fasi di sviluppo successive. Nel primo confronto a coppie (S1 vs S2), il “fenilpropanoide b.” era la via più altamente differenziata regolata, seguita da “amido e saccarosio m.” (Fig. 4). Un numero molto elevato di DEG (>100) è stato identificato in entrambe le vie. Un risultato simile è stato osservato nel seguente confronto a coppie, S2 vs. S3, ma nonostante il numero abbastanza elevato di DEG (>80) ‘amido e saccarosio m.’ha mostrato un valore q leggermente inferiore. Nel confronto S3 vs. S4, “la fissazione del carbonio negli organismi fotosintetici” era la via più significativamente regolata, ma il numero di geni era molto più basso. Nell’ultima coppia, S4 vs. S5,’ zeatin b.’, flavonoide b.’, acido grasso b. ‘e’ galattosio m. ‘ erano i percorsi più significativamente regolati, ma nessuno dei percorsi esibiva più di due DEG.

Trascrittoma LB: arricchimento della via metabolica del KEGG interstadio a coppie. (A) Fase 1 vs. fase 2. B) Fase 2 vs. fase 3. (C) Fase 3 vs. fase 4. D) Fase 4 contro fase 5. Top 15 (o tutti se totale <15) percorsi significativamente arricchiti sono mostrati. Il colore della barra rappresenta la grandezza del valore q (un valore p regolato da FDR). Tabella di colore è mostrato in figura.
LB Metabolome
Abbiamo condotto confronti di fase di sviluppo a coppie per identificare l’arricchimento dei metaboliti tra tutte le coppie di stadi. Il numero di metaboliti differenzialmente regolati nei confronti a coppie degli stadi di sviluppo in LB variava da 66 (stadi 3 vs 4) a 129 (stadi 2 vs 5). Il maggior numero nei confronti di stadi successivi è stato osservato tra il 1 ° e il 2 ° stadio (117). Questi sono stati assegnati a un gran numero di vie; con il maggior numero di metaboliti assegnati a ” b. dei metaboliti secondari “(anche il valore p più basso), seguito da “digestione e assorbimento delle proteine” e “b. degli amminoacidi” (Fig. 5; Cifre supplementari). Nella fase successiva confronto coppia (S2 vs. S3), “ABC transporters” è stata la via più significativamente arricchita, seguita da ” purine m.”. Nel confronto tra le coppie S3 e S4, “microbial m. in diversi ambienti” è stato il percorso più significativamente arricchito, seguito da ” carbapenem m.”. Nel confronto tra S4 e S5, un numero relativamente elevato di percorsi ha mostrato risultati simili (due metaboliti e valori p simili), ma notevole è la comparsa di “isoflavonoide b.” e “flavonoide b.” tra questi (Fig. 5; Cifre supplementari).

LB dati metabolomici: a coppie interstadio KEGG arricchimento via metabolica. (A) Fase 1 vs. fase 2. B) Fase 2 contro fase 3. (C) Fase 3 vs. fase 4. D) Fase 4 contro fase 5. Il fattore ricco è il rapporto tra il numero di metaboliti significativamente regolati nella via e il numero totale di metaboliti annotati in quella via (intervallo = da 0 a 1,0). La dimensione del punto rappresenta il numero di metaboliti significativamente arricchiti nella via corrispondente e il colore del punto rappresenta il valore P (entrambe le legende mostrate nella figura).
Per quanto riguarda i singoli metaboliti, nel confronto S1 vs. S2, l’elenco dei metaboliti sovraregolati nell’S1 è stato completato da trealosio, Glattinolo e acido L-malico (tutti ≈20 log2FC). L’acido oleico, l’acido 2-ossoadipico e l’acido stearico erano i metaboliti più altamente sovraregolati in S2 (tutti ≈15-18 log2FC). L’elenco dei metaboliti più altamente sovraregolati in S2 rispetto a S3 è stato superato da diidrossiacetone, LisoPC (18: 1 (9Z)) e Adenina (tutti ≈16 a 17,5 log2FC). L’elenco dei metaboliti più altamente sovraregolati nel S3 (rispetto a S2) è stato sormontato da trealosio, galattinolo e acido L-malico (tutti ≈19 a 20 log2FC). Nel confronto S3 vs. S4, l’elenco dei metaboliti più altamente sovraregolati nell’S3 era sormontato da acido L-Malico, DL-Arginina e acido oleico (tutti ≈18-19 log2FC), mentre l’elenco dei metaboliti più altamente sovraregolati nell’S4 era sormontato da D-Mannosio, N-Acetillattosamina e LisoPC(18:1(9Z)) in LB (tutti ≈16 log2FC). Nella S4 vs. Confronto S5, l’elenco dei metaboliti più altamente upregulated nel S4 è stato sormontato da trealosio, acido palmitico, N-Acetyllactosamine (tutti ≈17 a 18 log2FC) in LB. L’elenco dei metaboliti più altamente sovraregolati nel S5 è stato completato da L-Norleucina, acido antranilico (vitamina L1) e DL-Arginina (tutti ≈16 a 18 log2FC) (Set di dati supplementari S3).
Lycium ruthenicum (LR): analisi interstadiali a coppie
Trascrittoma LR
Heatmap l’analisi dei DEG in LR mostra che insiemi quasi completamente diversi di geni sono stati altamente sovraregolati nelle prime fasi dello sviluppo (1 + 2) e nella frutta matura (stadio 5), con un apparente reset trascrittomico che si verifica dopo il secondo stadio (Fig. 3 TER). L’analisi della parentela del campione indica che i campioni potrebbero essere divisi in due cladi (fasi 1 + 2 e 3 + 4 + 5), con quest’ultimo clade ulteriormente suddiviso in due cladi: fasi 3 + 4 e fase 5. L’analisi di classificazione funzionale intraspecifica di KEGG di questi DEG ha identificato 35 percorsi in modo significativo (P < 0.05) differenzialmente regolati tra diversi stadi di sviluppo (Set di dati supplementari S2). Particolarmente fortemente differenzialmente regolati erano ‘fotosintesi’, ‘ acido linoleico m.’, ‘ alcaloide isochinolina b.’, ‘flavonoide b.’ e ‘ amido e saccarosio m ‘(Fig. 3 TER). Il numero più alto di DEG è stato identificato nella maggior parte dei confronti a coppie dello stadio 1 (1 contro 2 era un’eccezione) e nel confronto dello stadio 2 contro 5 (tutti >5.000 DEG; Tabella 2). Di gran lunga il numero più piccolo è stato identificato nel confronto S3 vs. S4 (39). Nel confronto a coppie S1 vs S2, “amino sugar and nucleotide sugar m.” era la via più altamente differenziata regolata, seguita da “linoleic acid m.” (Fig. 6). Nel confronto a coppie S2 vs. S3, le vie più significativamente differenziate sono state ” alcaloide isochinolina b.”, ” tirosina m.”, “fenilpropanoide b” e ” flavonoide b.”. In entrambi i confronti a coppie, il più alto numero di DEG (>15 e >35 rispettivamente) è stato identificato nel ” amido e saccarosio m.”. Nella S3 vs. Confronto S4, ‘(alfa-)acido linoleico m. ‘ era la via più significativamente regolata, ma il numero di geni erano molto più bassi. Nel confronto S4 vs S5, “fenilpropanoide b” (anche il maggior numero di DEG) e “acido linoleico m.” sono stati i percorsi più significativamente regolati.
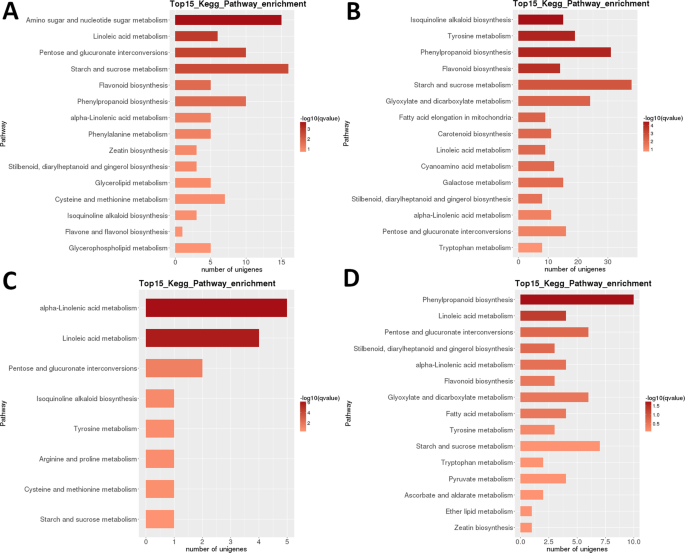
Trascrittoma LR: arricchimento della via metabolica del KEGG interstadio a coppie. (A) Fase 1 vs. fase 2. B) Fase 2 contro fase 3. (C) Fase 3 vs. fase 4. D) Fase 4 contro fase 5. Top 15 (o tutti se totale <15) percorsi significativamente arricchiti sono mostrati. Il colore della barra rappresenta la grandezza del valore q (un valore p regolato da FDR, grafico a colori mostrato in figura).
Metaboloma LR
Il numero di metaboliti differenzialmente regolati nei confronti a coppie degli stadi di sviluppo della LR variava da 66 (stadi 3 vs 4) a 133 (stadi 2 vs 5) (Dataset supplementare S3). Nei confronti di stadi successivi, il maggior numero è stato osservato tra il 1 ° e il 2 ° stadio (117). Questi sono stati assegnati a un gran numero di percorsi; con il maggior numero di metaboliti assegnati a “b dei metaboliti secondari”, seguito da “digestione e assorbimento delle proteine”, “b. degli amminoacidi” e “flavonoide b”. Nel confronto S2 vs. S3 ,’ ABC transporters ‘è stato il percorso più significativamente arricchito, seguito da’purine m’. Nel confronto tra le coppie S3 e S4, “microbial m. in diversi ambienti” è stato il percorso più significativamente arricchito, seguito da ” carbapenem m.”. Nella S4 vs. Confronto delle coppie S5, un numero relativamente elevato di percorsi ha mostrato risultati simili (2 metaboliti e valori p simili), ma notevole è la comparsa di “isoflavonoide b.” e “flavonoide b.” tra questi (Fig. 7; Cifre supplementari).

Dati metabolomici LR: arricchimento della via metabolica di KEGG interstadio a coppie. (A) Fase 1 vs. fase 2. B) Fase 2 contro fase 3. (C) Fase 3 vs. fase 4. D) Fase 4 contro fase 5. Il fattore ricco è il rapporto tra il numero di metaboliti significativamente regolati nella via e il numero totale di metaboliti annotati in quella via (intervallo = da 0 a 1,0). La dimensione del punto rappresenta il numero di metaboliti significativamente arricchiti nella via corrispondente e il colore del punto rappresenta il valore P (entrambe le legende mostrate nella figura).
Per quanto riguarda i singoli metaboliti (dataset supplementare S3), nel S1 vs. Confronto S2, l’elenco dei metaboliti sovraregolati nel S1 è stato sormontato da trealosio, galattinolo e acido L-malico (≈19-21 log2FC), mentre l’acido oleico, l’acido 2-ossoadipico e l’acido stearico erano i metaboliti più altamente sovraregolati in S2 (≈15-18 log2FC). Nel confronto S2 vs. S3, Diidrossiacetone, Indoxyl sulphate e N-Acetyllactosamine sono stati metaboliti più altamente sovraregolati nel S2 (≈17-19.5 log2FC), e trealosio, galattinolo e acido L-malico (≈19-21 log2FC) nel S3. Nella S3 vs. Confronto S4, acido L-malico, DL-arginina e acido oleico sono stati sovraregolati nella S3 (≈16-19 log2FC) e 1,7-dimetilxantina, D-mannosio e N-Acetillattosamina (≈15-17 log2FC) nella S4. Nel confronto S4 vs. S5, Flavin mononucleotide, trealosio e acido isoferulico sono stati upregolati nel S4 (≈18-20 log2FC), mentre PG(16:0/18:1(9Z)), D-Prolina e DL-Arginina sono stati i metaboliti più altamente upregolati (tutti ≈16-18 log2FC).
Analisi comparativa interspecifica di DEGs in diverse fasi di sviluppo
DEGs totali durante lo sviluppo del frutto
Confronto interspecifico a coppie (LR1 vs. LB1, LR2 vs. LB2, ecc.) mostra che 928 DEG erano condivisi da tutte e cinque le coppie (Fig. 8 BIS). Il più alto numero di DEG è stato identificato nella fase 3 (3989) e il più basso nella fase 4 (2825) (Fig. 8B); mentre il numero più alto di DEG unici per una coppia è stato osservato negli stadi 3 (574), 1 e 5 (entrambi 554) e il più basso nello stadio 4 (126) (Fig. 8 BIS). I numeri di DEG regolati su e giù erano relativamente simili in ciascuno dei confronti di fase a coppie; ad esempio, nella fase 5, 1668 DEG erano sovraregolati e 1670 DEG erano downregolati in LR rispetto a LB (Fig. 8 TER). Tuttavia, negli altri quattro stadi il numero di geni sovraregolati era leggermente più alto (da 93 a 189 DEG).

Geni espressi in modo differenziale (DEGs) tra i frutti di L. barbarum (LB) e L. ruthenicum (LR). (A) Comparazioni dettagliate a tappe (da 1 a 5) (LB vs. LR). (B) Il numero di DEGS upregulated (rosso) e downregulated (verde) in LR rispetto a LB in cinque fasi di sviluppo studiate.
Transcriptome-pathways
Heatmap l’analisi dei DEG indica che i frutti di due specie presentano profili di espressione genica molto diversi durante tutte le fasi dello sviluppo, ma le repliche biologiche hanno mostrato profili molto simili, indicando una quantità limitata di variabilità individuale in ogni fase dello sviluppo (Figure supplementari: Fig. S9). L’analisi comparativa dell’arricchimento della via di KEGG mostra che solo alcune vie erano costantemente altamente arricchite (in termini di regolazione genica) in LR rispetto a LB durante tutte e cinque le fasi dello sviluppo (Fig. 9). In particolare, la trasduzione del segnale dell’ormone vegetale (2a-più alta in S1, 8a-più alta in S2, la più alta in S3, S4 e S5) e l’interazione pianta-patogeno (la più alta in S1, 3a-più alta in S2, 2a-più alta in S3, 4a-più alta in S4 e 15a – La biosintesi del fenilpropanoide (non nella top 15 in S1, la più alta in S2, la 3a-più alta in S3, la 7a-più alta in S4, la 11a-più alta in S5), l’ubichinone e altre biosintesi del terpenoide-chinone (non nella top 15 in S1, la 6a-più alta in S2, la 7a-più alta in S3, la 2a-più alta in S4, la 6a-più alta in S5) erano anche relativamente altamente sovraregolate in tutte le fasi tranne la prima. La via della biosintesi dei flavonoidi non è stata altamente arricchita nelle fasi iniziali (non nella top 15 in S1, 14th-highest in S2) e altamente arricchita nelle fasi successive (3rd to 4th-highest durante le fasi da 3 a 5). (alfa-)Il metabolismo dell’acido linoleico è stato altamente arricchito negli stadi intermedi (9 ° -più alto in S1, 2 ° -più alto in S2, 5 ° e 6 ° -più alto in S3, 8 ° -più alto in S4, non in top 15 in S5).

Analisi comparativa dell’arricchimento della via metabolica del KEGG. I primi 15 percorsi arricchiti in L. ruthenicum rispetto a L. barbarum sono mostrati a sinistra (rosso), e quelli arricchiti in L. barbarum rispetto a L. ruthenicum a destra (verde). Le fasi di sviluppo (1-5) sono indicate nella figura. il valore q è un valore p regolato da FDR.
Tra i percorsi downregulated in L. ruthenicum rispetto a L. barbarum (Fig. 9) sono stati osservati cambiamenti notevoli tra le fasi iniziali (1 e 2), quando il metabolismo degli acidi cianoammino e la biosintesi dei carotenoidi erano più fortemente downregolati, e le fasi successive (4 e 5), quando le interazioni RULLANTI nel trasporto vescicolare, il metabolismo di nicotinato e nicotinammide e il metabolismo di porfirina e clorofilla erano costantemente relativamente altamente downregolati.
Trascrittoma – geni individuali
Tra i geni più altamente differenzialmente espressi, alcuni erano specifici dello stadio dello sviluppo (cioè altamente differenzialmente regolati solo nelle fasi iniziali o tardive dello sviluppo), ma alcuni erano costantemente altamente differenzialmente regolati in tutte e cinque le fasi studiate (Set di dati supplementare S4). Diversi geni legati all’immunità molto altamente sovraregolati in LR rispetto a LB nelle prime fasi dello sviluppo sono tra gli esempi del modello di espressione specifico dello stadio dello sviluppo: la chitinasi era il 2 ° GRADO più alto sovraregolato in S1 (13,43 volte), il più alto in S2 (13,89 volte), ma nelle fasi successive non era un GRADO. Allo stesso modo, il recettore EIX 1/2 è stato anche molto sovraregolato nelle fasi iniziali, il più alto in S1 (13.70) e 2nd-highest in S2 (10.90), ma non è stato identificato come DEG nelle fasi successive. Alcuni geni correlati alla crescita hanno anche mostrato un modello di espressione simile: la fosfoglicerato chinasi (PGK) era tra la manciata di geni più altamente sovraregolati nei primi tre stadi (13.14, 12.87 e 12.77 rispettivamente), ma non è stato identificato come DEG nelle fasi successive. Anche la subunità complessa CCR4-NOT transcription 7/8 (CNOT7/8) ha mostrato un modello di espressione molto simile: altamente sovraregolato nei primi tre stadi e non un GRADO negli stadi 4 e 5. Diversi geni associati alla biosintesi di flavonoidi e fenilpropanoidi hanno mostrato un modello di espressione specifico dello stadio dello sviluppo invertito, con un’espressione relativamente bassa nelle fasi iniziali e molto alta nelle fasi successive. Esempi sono: bifunzionale diidroflavonolo 4-reduttasi/flavanone 4-reduttasi (DFR), che è stato leggermente sovraregolato in LR in S1 (2.25), non un GRADO in S2, altamente upregulated in S3 (7.79), e 3rd-più alto gene upregulated in S4 (14.25) e S5 (16.03). Un paralogo di questo gene ha mostrato un modello quasi identico: leggermente sovraregolato in S1 (2.44), non un GRADO in S2, altamente sovraregolato in S3 (7.40), 6 ° -più alto GRADO sovraregolato in S4 (13.26) e 5 ° in S5 (14.59). Allo stesso modo,il flavonoide 3′, 5′-idrossilasi (F3’5’H) non era un GRADO nei primi due stadi, altamente sovraregolato in S3 (6.69), 5 ° -più alto gene sovraregolato in S4 (13.42) e 4 ° -più alto in S5 (15.05). La flavonoide O-metiltransferasi (OMT) non era un GRADO in S1, ma in S2 mostrava già un livello di upregulation medio-alto (4.32), da S3 era già il terzo più alto grado di upregulated (13.30), ed era il gene più alto-upregulated in S4 (18.73) e S5 (18.10). La leucoantocianidina diossigenasi (LDOX; biosintesi antocianica) non era un GRADO in S1 e S2, seguita da un aumento da alto a molto alto nelle fasi successive (5.63, 9.44, 11.56, rispettivamente). Anche due paraloghi calcone sintasi (CHS e CHS2; biosintesi flavonoide) non erano altamente regolati in S1 e S2 (CHS2: non a DEG, CHS: -1.14 in S1, non un GRADO in S2), ma in S3–S5 entrambi i geni hanno mostrato una sovraregolazione medio-alta (CHS2: 5.32, 7.84, 6.00; e CHS: 4.67, 7.01, 6.82; rispettivamente). Abbiamo selezionato questi geni per l’analisi qPCR e i risultati sono altamente congruenti con i dati RNA-seq (Risultati supplementari; Set di dati supplementari S5). Infine, la citochinina deidrogenasi, un gene correlato alla biosintesi della zeatina, è stata sempre più sovraregolata durante gli ultimi tre stadi (2.6–5.7).
Tuttavia, alcuni geni sono stati espressi in modo coerente e differenziato in tutte e cinque le fasi studiate. Esempi includevano anche alcuni geni correlati all’immunità, come due paraloghi di glutatione S-transferasi, altamente sovraregolati in LR rispetto a LB in tutte le fasi: 9.38 e 8.58 (tutti i valori presentati come cambiamenti di piega nel rispettivo ordine) in S1, 6.30 e 6.34 in S2, il 2 ° e 7 ° più alto DEGS sovraregolato in S3 (14.08 e 12.70), 14.16) e 2 ° e 6 ° più alto in S5 (16.48 e 14.40). Anche la proteina di resistenza alle malattie delle piante RPM1 è stata altamente sovraregolata in tutte e cinque le fasi (S1 = 13.15; S2 = 12.08; S3 = 13.11, S4 = 12.81; S5 = 13:94). Tra i geni espressi in modo coerente e differenziato in tutte le fasi dello sviluppo c’erano anche alcuni correlati al metabolismo degli aminoacidi, ma il loro modello è stato invertito rispetto agli esempi precedenti: hanno mostrato un’alta downregulation in LR rispetto a LB. Esempi sono acetil-CoA aciltransferasi 1 (AAT1; degradazione di valina, leucina e isoleucina), con un profilo temporale di downregulation sempre più elevato, a partire da -7.0 nel S1 a <−10 volte negli ultimi tre stadi. La prolina iminopeptidasi, associata all’arginina e al metabolismo della prolina, è stata altamente ridotta nella LR in tutte le fasi: S1 = -9,75, S2 = -10,89 (3 ° -più alto), S3 = -11,05 (4 ° -più alto), S4 = -10,01 e S5 = -11,98 (3 ° -più alto). Infine, la 5–metiltetraidropteroiltriglutammato-omocisteina metiltransferasi (metE) è stata costantemente estremamente ridotta in LR in tutte le fasi: 2a-più alta in S1 (-11.76), la più alta in S2 (-11.74), 3a-più alta in S3 (-11.43), la più alta in S4 (-12.36) e 2a-più alta in S5 (-12.83). Anche due geni associati alla replicazione del DNA e alla trascrizione sono stati fortemente downregolati in LR in tutte le fasi: la proteina nucleare legante GTP Ran (RAN; da -10,0 a -12,0) e il fattore di replicazione A1 (RFA1) (da -8,0 a -12,0). Alcuni geni correlati alla crescita e allo stress sono stati anche costantemente fortemente downregolati in LR: ribonucleoproteina nucleare eterogenea A1/A3 (hnRNP; da -7 a -11) e shock termico 70 kDa proteina 1/8 (HSPA1_8) S1 = -4.95, S2 = -8.88, S3 = -11.48 (2nd-highest), S4 = -9.36, S5 = -12.89 (the highest). Curiosamente, un gene correlato alla biosintesi fenilpropanoide, shikimate hydroxycinnamoyltransferase (HCT), è stato anche costantemente altamente downregulated in LR: S1 = -6.82, S2 = -8.14, S3 = -11.71 (il più alto), S4 = -11.00 (3rd-highest), S5 = -11.91 (4th-highest). Tuttavia, un regolatore chiave della biosintesi dell’antociano, il fattore di trascrizione MYB114, è stato altamente sovraregolato in LR durante tutte e cinque le fasi di sviluppo: 6.11, 4.69, 7.47, 9.05 e 8.95 (S1–S5 rispettivamente).
Metabolome – pathways
Abbiamo anche condotto un’analisi comparativa interspecifica delle vie metaboliche (Fig. 10). Nella prima fase di sviluppo (S1), abbiamo identificato 39 metaboliti differenzialmente regolati. Tra i primi 20 percorsi a cui questi metaboliti erano associati, molti di essi erano associati agli amminoacidi, ma il numero totale di metaboliti per via era relativamente piccolo (1-2) e i valori di P non suggerivano un alto livello di significatività (Fig. 10-pannello 1). In particolare il metabolismo della vitamina B6 (m) e il m microbico in diversi ambienti hanno entrambi mostrato valori P relativamente alti, fattore di arricchimento (EF) di 1,0 e 2 metaboliti identificati. Nel S2, abbiamo identificato 58 metaboliti differenzialmente regolati, associati a solo quattro vie: triptofano m, biosintesi dei fenilpropanoidi (b), b dei fenilpropanoidi (questi sono due percorsi diversi nel database di KEGG) e fenilalanina, tirosina e triptofano b (tutti i metaboliti EF = 1.0, 2-3 e p < 0.5; Fig. 10-pannello 2). Nel S3, abbiamo identificato 59 metaboliti differenzialmente regolati, associati a 19 vie, la maggior parte tutte con EF 1.0, ma valori P relativamente non significativi (>0.5; Fig. 10-pannello 3). Le vie con un numero relativamente elevato di metaboliti (n = 5) erano: digestione e assorbimento delle proteine, b dei metaboliti secondari vegetali, b degli antibiotici e b degli amminoacidi. Nel S4, abbiamo identificato 58 metaboliti differenzialmente regolati, associati a un gran numero di vie, per lo più con l’EF 1.0, e valori di significatività relativamente alti (principalmente P > 0.5; Fig. 10-pannello 4). Percorsi con un numero relativamente alto di metaboliti (n ≥ 3) sono stati: fenilpropanoide b, fenilalanina, tirosina e triptofano b, glucosinate b, b di alcaloidi derivati da shikimato percorso, e 2-oxocarboxylic m. La frutta matura (S5), abbiamo individuato 39 differenzialmente metaboliti, associato con un gran numero di percorsi, ma soprattutto con bassi valori di P e solo 1 metabolita al percorso (Fig. 10-pannello 5). Le vie con più di 1 metabolita sono state: digestione e assorbimento delle proteine, fenilpropanoide b, assorbimento dei minerali, carbonio centrale m nel cancro, b dei metaboliti secondari, b dei fenilpropanoidi e aminoacy tRNA b. L’analisi dei componenti principali (PCA) di tutti i dati (2 specie × 5 stadi × 5 repliche biologiche) ha rivelato un’elevata somiglianza tra le repliche biologiche (clustering) e ha confermato una notevole variabilità tra 10-pannello 6).

Analisi comparativa dell’arricchimento della via metabolica del KEGG. I primi 15 percorsi arricchiti in L. ruthenicum rispetto a L. barbarum sono mostrati a sinistra (rosso), e quelli arricchiti in L. barbarum rispetto a L. ruthenicum a destra (verde). Le fasi di sviluppo (1-5) sono indicate nella figura. il valore q è un valore p regolato da FDR.
Metaboloma-singoli metaboliti
L ‘ elenco (Dataset supplementare S6) dei metaboliti più differenzialmente regolati tra le due specie, ha mostrato alcune variazioni tra le cinque fasi di sviluppo del frutto. Curiosamente, il fruttosio 1-fosfato è stato il metabolita più altamente sovraregolato in LR, rispetto a LB, durante tutte e cinque le fasi: log2 Fold change = 6.3, 7.6, 7.7, 8.1 e 6.5 (stadi da 1 a 5 rispettivamente). Per quanto riguarda i metaboliti altamente upregulated in LB, c’era più variazione fra le fasi: nel S1, le differenze erano piuttosto piccole, con il 9-Decenolo come metabolita più altamente sovraregolato (log2 Fold change = 2.7; rispetto a LR). Le analisi di S2 e S3 hanno prodotto risultati altamente congruenti, con fenolo (3.7 e 3.2 rispettivamente) come metabolita più altamente sovraregolato. Nel S4, l’indossilsolfato (4.7) era il principale metabolita sovraregolato in LB. Un cambiamento metabolico è stato osservato nel frutto maturo (S5), dove l’elenco dei metaboliti upregulated in LB è stato sormontato da stearoilcarnitina (7.1), acido metossiacetico (5.3), S-Metil-5′-tioadenosina (4.7), lisinopril (4.7), Adenosina 3’, 5 ‘ -fosfato ciclico (cAMP) (4.7), ecc. Altri metaboliti altamente sovraregolati nella LR (a parte il Fruttosio 1-fosfato) erano naringina (6,2), lauroil-CoA (4,8), L-Phneylalanina (4,6), ecc.