La presentazione clinica più comune dell’iperparatiroidismo primario (PHP) è l’ipercalcemia asintomatica e la diagnosi di PHP basata sulla presenza di manifestazioni ossee come l’osteite fibrosa cistica (OFC) è sempre più rara. OFC si verifica in meno del 5% dei pazienti con PHP e suggerisce una malattia più grave o di lunga data. OFC è caratterizzata dalla presenza di dolore osseo associato con il ritrovamento di cambiamenti radiografici specifici come un aumento riassorbimento osseo subperiostale nel terzo distale del radio e falangi medie, distale assottigliamento clavicolare, “sale e pepe” cranio, cisti ossee, e tumori marroni nelle ossa lunghe. I tumori marroni derivano dalla demineralizzazione ossea con attivazione osteoclastica, microemorragie e microfratture e sono così chiamati a causa del loro colore tipico, a causa di abbondanti depositi di emosiderina. Istopatologicamente, esiste una combinazione di attività osteoclastica e osteoblastica con formazione di cisti e molti macrofagi carichi di emosiderina.1 La diagnosi differenziale dei tumori marroni comprende il granuloma riparativo a cellule giganti e il tumore a cellule giganti (GCT) dell’osso.
È riportato il caso di un paziente con PHP a causa di un adenoma paratiroideo con tumori marroni che imitano un GCT metastatico.
Un 47enne ha frequentato per la prima volta il reparto di chirurgia ortopedica di un ospedale in Castiglia-La Mancha nel maggio 2008 lamentando dolore all’anca e alla mano sinistra non associati a traumi precedenti. Il paziente ha riportato una storia personale di dislipidemia, diabete mellito di tipo 2, ipertensione arteriosa, obesità di grado I e colica renale con calcoli di ossalato di calcio. La sua storia familiare comprendeva due figlie che erano state diagnosticate e sottoposte a un intervento chirurgico per PHP a causa di adenoma. La radiografia semplice dell’anca e della mano ha mostrato un’immagine cistica polilobulata che ha gonfiato e assottigliato l’osso corticale nel terzo osso metacarpale sinistro e le lesioni litiche del ramus suproischiopubico sopra-acetabolare e sinistro. Una TAC del bacino (novembre 2008) ha mostrato grandi lesioni nell’ala iliaca, nell’ischio, nel ramus pubico sinistro, nell’ala sacrale destra e nel collo del femore. Sulla base di questi risultati, nel mese di luglio e settembre 2009 il paziente è stato sottoposto a un intervento chirurgico consistente in curettage e riempimento sia con un innesto autologo che con sostituti ossei del terzo metacarpo sinistro e della lesione sopracetabolare sinistra. Il laboratorio patologico ha riportato un GCT. Controlli successivi con TC e RM del torace e del bacino hanno rivelato l’allargamento delle lesioni litiche polilobulate ed espansive nel bacino (Fig. 1), sacro, collo femorale destro e L5, con la comparsa di una nuova lesione nella testa femorale destra e nel settimo arco costale sinistro (Fig. 2). Questi cambiamenti sono stati attribuiti alla progressione del tumore metastatico. A causa del dolore persistente che impediva totalmente la deambulazione, il paziente è stato indirizzato all’unità tumorale ossea dell’Ospedale Universitario La Paz, dove una revisione di campioni patologici ha portato alla conclusione che le lesioni ossee erano altamente suggestive di granulomi riparativi a cellule giganti e istologicamente indistinguibili da tumori marroni. L’iperparatiroidismo è stato quindi escluso. Nel novembre 2010, il paziente è stato indirizzato al reparto di endocrinologia, dove ulteriori test di laboratorio hanno fornito i seguenti risultati: calcio totale 14 mg/dL, calcio corretto 13,2 mg/dL, calcio ionico 1,72 mmol/L, fosfato 1,9 mg/dL, magnesio 1,86 mg/dL, calcio urinario 968,60 mg/24h, creatinina 0,55 mg/dL, iPTH 535pg/mL, vitamina D 13ng/mL. La TC total body ha mostrato un nodulo di 1,5 cm di diametro nella posizione teorica della ghiandola paratiroidea destra e una scansione paratiroidea con 20mCi di TC 99-sestamibi ha rivelato risultati coerenti con l’adenoma paratiroideo iperfunzionante destro. Inoltre, la presenza di un feocromocitoma associato è stata esclusa in base sia alla biochimica che alla morfologia. È stata eseguita la paratiroidectomia destra e una biopsia intraoperatoria è stata segnalata come adenoma paratiroideo. Lo studio istopatologico finale ha confermato la presenza di un adenoma paratiroideo di 4,5 g di peso e di 2,2 cm×2 cm×1,9 cm di dimensione.
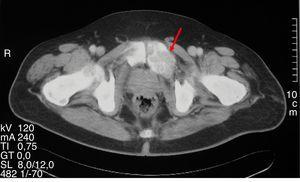
Figura 1.
Lesione litica espansiva corrispondente all’osteite fibrosa cistica in una TAC pelvica.
(0.14 MB).
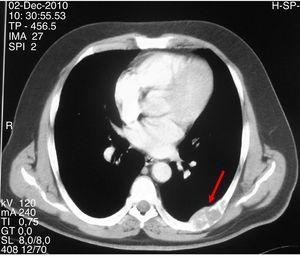
Figura 2.
TAC che mostra una lesione litica espansiva con contorno lobulato e assottigliamento corticale nel settimo arco costale sinistro.
(0.21 MB).
Dopo l’intervento chirurgico, il paziente ha avuto ipocalcemia sintomatica che ha richiesto un trattamento con calcitriolo e calcio, che è stato continuato fino ad oggi. Continua ad essere seguito presso il dipartimento di endocrinologia, riporta un significativo miglioramento sintomatico ed è in grado di camminare con le stampelle. Uno studio genetico non ha trovato mutazioni nel gene MEN-1.
GCT è un tumore altamente vascolarizzato che si trova nelle metafisi o epifisi delle ossa lunghe o nel bacino, sacro o vertebre.2 L’aspetto radiografico e istologico dei tumori bruni tipici dell’OFC può imitare da vicino un GCT, come si è verificato nel nostro paziente, e la differenziazione dovrebbe essere fatta sulla base di segni clinici e risultati di laboratorio (iPTH). Alcuni degli autori3, 4 hanno riportato casi di OFC in cui la malattia ossea metastatica secondaria è stata inizialmente sospettata sulla base di segni clinici e immagini radiografiche. Nel nostro paziente, tuttavia, la diagnosi di un tumore osseo primario metastatico era stata basata su risultati istologici, mentre la storia familiare, non riportata nei casi precedenti, era coerente con PHP.
D’altra parte,la carenza di vitamina D è spesso rilevata in pazienti con PHP e is3, 5 associati ad esacerbazione della presentazione biochimica e fenotipica della malattia (livelli di PTH sierico più elevati, adenomi paratiroidei di grandi dimensioni e maggiore rischio di frattura), che possono aver contribuito al quadro clinico florido del nostro paziente.
Le forme familiari di iperparatiroidismo sono note per essere non comuni (5%) e le loro cause più frequenti includono sindromi multiple di neoplasia endocrina (MEN) di tipo 1 e 2A, sindrome da tumore della mascella iperparatiroidismo (HPT-JT) e iperparatiroidismo isolato familiare (FIHP).6 Negli UOMINI 1, l’iperparatiroidismo è la presentazione più precoce e più comune (>90%), mentre negli UOMINI 2A si verifica in ritardo e ha una bassa penetranza. Sebbene lo studio genetico per MEN 1 fosse negativo nel nostro paziente, va notato che un risultato falso negativo può verificarsi fino al 30% dei casi testati come risultato di modelli di mutazione che coinvolgono diverse regioni geniche o mutazioni in geni ancora sconosciuti che influenzano la trascrizione o l’azione di menin.7 Questo, insieme al probabile verificarsi asincrono di diversi aspetti di MEN-1, rende necessario un monitoraggio continuo. MEN 2A è improbabile in assenza di coinvolgimento neoplastico tiroideo o feocromocitoma. La diagnosi differenziale dovrebbe includere anche HPT-JT a causa della scala del coinvolgimento osseo e delle grandi dimensioni dell’adenoma. La scoperta finale di un carcinoma paratiroideo avrebbe supportato questa diagnosi, a causa della sua frequente presenza in HPT-JT.8 Tuttavia, l’assenza di lesioni fibro-ossee mandibolari o mascellari e lesioni renali lo ha reso improbabile. Infine, sebbene la FIHP possa rappresentare in alcuni casi una variante di altre sindromi iperparatiroidee, la possibilità che mutazioni localizzate in loci non ancora identificati diversi da quelli riportati negli UOMINI 1 e 2 e nell’HPT-JT possano causare questa sindrome non può essere esclusa.
L’interesse del caso riportato risiede nel fatto che illustra l’importanza di valutare il metabolismo del fosforo e del calcio e la funzione paratiroidea in tutti i pazienti con lesioni ossee, di sospettare un potenziale PHP se esistono lesioni suggestive e di cercare una probabile componente genetica sottostante attraverso una dettagliata storia familiare e personale.