Illumina sekvensering teknologi bruker cluster generasjon og sekvensering av syntese (sbs) kjemi til sekvens millioner eller milliarder av klynger på en strømningscelle, avhengig av sekvensering plattform. Under sbs kjemi, for hver klynge, base samtaler er laget og lagret for hver syklus av sekvensering Av Real-Time Analyse (RTA) programvare på instrumentet. RTA lagrer base call data i form av individuelle base call (ELLER BCL) filer. Når sekvensering er fullført, må baseanropene I BCL-filene konverteres til sekvensdata. DENNE prosessen kalles BCL TIL FASTQ konvertering.
EN FASTQ-fil er EN tekstfil som inneholder sekvensdataene fra klyngene som sender filteret på en flytcelle (hvis du vil ha mer informasjon om klynger som sender filteret, kan du se delen «tilleggsinformasjon» i denne bulletinen). Hvis prøvene ble multipleksert, er det første trinnet I FASTQ filgenerering demultiplexing. Demultiplexing tilordner klynger til en prøve, basert på klyngen indeks sekvens (r). Etter demultiplexing skrives de samlede sekvensene TIL FASTQ-filer per prøve. Hvis prøver ikke ble multipleksert, forekommer ikke demultipleksingstrinnet, og for hver flytcellefelt tilordnes alle klynger til en enkelt prøve.
for en enkeltlesning, opprettes EN LES 1 (R1) FASTQ-fil for hver prøve per flytcellefelt. For en sammenkoblet sluttkjøring opprettes en R1 OG En Read 2 (R2) FASTQ-fil for hver prøve for hver kjørefelt. FASTQ-filer komprimeres og opprettes med utvidelsen *.fastq.gz.
hvordan ser EN FASTQ-fil ut?
for hver klynge som passerer filter, skrives en enkelt sekvens Til Den tilsvarende prøvens R1 FASTQ-fil, og for en sammenkoblet sluttkjøring skrives en enkelt sekvens også Til prøvens R2 FASTQ-fil. Hver oppføring I EN FASTQ filer består av 4 linjer:
- en sekvensidentifikator med informasjon om sekvensering kjøre og klyngen. Det nøyaktige innholdet i denne linjen varierer fra BASERT PÅ BCL TIL FASTQ-konverteringsprogramvaren som brukes.
- sekvensen (basen kaller; A, C, T, G og N).
- en separator, som bare er et pluss ( + ) tegn.
- de grunnleggende anropskvalitetspoengene. Disse Er Phred + 33 kodet, VED HJELP AV ASCII-tegn for å representere de numeriske kvalitetspoengene.
her er et eksempel på en enkelt oppføring I EN R1 FASTQ-fil:
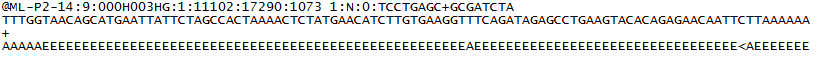
Mer detaljert informasjon OM FASTQ-formatet finner du her.
slik viser DU EN FASTQ-fil
FASTQ-filer kan inneholde opptil millioner av oppføringer og kan være flere megabyte eller gigabyte i størrelse, noe som ofte gjør dem for store til å åpne i en vanlig tekstredigerer. VANLIGVIS er DET ikke nødvendig å vise FASTQ-filer, fordi de er mellomliggende utdatafiler som brukes som input for verktøy som utfører nedstrømsanalyse, for eksempel justering til en referanse eller de novo-samling.
hvis DU trenger Å se EN FASTQ-fil for feilsøkingsformål eller av nysgjerrighet, trenger du enten en tekstredigerer som kan håndtere svært store filer, eller tilgang Til Et Unix-eller Linux-system hvor store filer kan vises via kommandolinjen.
slik genererer DU FASTQ-filer
FASTQ-filgenerering ER det første trinnet for alle analysearbeidsflyter som Brukes Av MiSeq Reporter På MiSeq og Local Run Manager på MiniSeq. NÅR analysen er fullført, LIGGER FASTQ-filene i < kjør mappe > \ Data \ Intensiteter \ BaseCalls På miseq og <output-mappen > \Alignment_ # \ < undermappe> \ Fastq På MiniSeq.
FOR alle kjøringer lastet opp Til BaseSpace Sequence Hub, OPPSTÅR FASTQ – filgenerering automatisk etter at kjøringen er fullstendig lastet opp, OG FASTQ-filene brukes som inngang for De ulike analyseappene På BaseSpace Sequence Hub. På BaseSpace Sequence Hub kan du finne FASTQ-filene DINE i prosjektet (e) som er knyttet til kjøringen din.
den bcl2fastq konvertering programvare kan brukes til å generere FASTQ filer fra data generert på alle aktuelle Illumina sekvensering systemer.
hvis du vil ha informasjon om de ulike innstillingene som KAN brukes under GENERERING AV FASTQ-filer, kan du se brukerveiledningene for programvaren nedenfor.
- MiSeq Reporter
Lokal Run Manager
bcl2fastq
Tilleggsinformasjon
- en beskrivelse og krav for klynger til pass filter kan bli funnet i seksjon 1.5.8 Av MiSeq: Imaging Og Base Calling online kurs.
- Se 2-Kanals SBS-Teknologi for mer informasjon om baseanrop På NovaSeq, NextSeq 500/550 og MiniSeq-systemer.
- Se Illumina Sekvensering Teknologi for mer informasjon om base ringer På MiSeq og HiSeq systemer.