de meest voorkomende klinische presentatie van primaire hyperparathyreoïdie (PHP) is asymptomatische hypercalciëmie, en de diagnose van PHP gebaseerd op de aanwezigheid van bot manifestaties zoals osteitis fibrosa cystica (OFC) wordt steeds zeldzamer. OFC komt voor bij minder dan 5% van de patiënten met PHP en suggereert een ernstigere of langdurige ziekte. OFC wordt gekenmerkt door het optreden van botpijn geassocieerd met het vinden van specifieke radiografische veranderingen zoals verhoogde subperiosteale botresorptie in het distale derde van de radius en Midden falanges, distale claviculaire dunner worden, “zout en peper” schedel, botcysten, en bruine tumoren in lange botten. De bruine tumors resulteren uit beendemineralisatie met osteoclastactivering, microhemorrhages, en microfracturen, en worden zo genoemd wegens hun typische kleur, wegens overvloedige hemosiderin afzettingen. Histopathologisch bestaat er een combinatie van osteoclastische en osteoblastische activiteit met cyste vorming en veel hemosiderine-geladen macrofagen.1 differentiële diagnose van bruine tumoren omvat giant cell reparative granuloma en giant cell tumor (GCT) van het bot.
het geval van een patiënt met PHP als gevolg van een bijschildklieradenoom met bruine tumoren die een gemetastaseerd GCT nabootsen wordt gerapporteerd.
een 47-jarige ging in Mei 2008 voor het eerst naar de afdeling orthopedische chirurgie van een ziekenhuis in Castilië-La Mancha en klaagde over pijn in de heup en linkerhand die niet geassocieerd was met eerder trauma. De patiënt meldde een persoonlijke geschiedenis van dyslipidemie, type 2 diabetes mellitus, arteriële hypertensie, graad I obesitas, en nierkoliek met calciumoxalaatstenen. Zijn familiegeschiedenis omvatte twee dochters die waren gediagnosticeerd en een operatie voor PHP als gevolg van adenoom. Röntgenfoto ‘ s van de heup en de hand toonden een polylobulated cystische afbeelding die het corticale bot in het derde linker metacarpale bot en supra-acetabulaire en linker ilioischiopubische ramus lytische laesies opgeblazen en verdund. Een CT-scan van het bekken (November 2008) toonde grote laesies in de iliacale ala, ischium, linker pubic ramus, en rechter sacrale vleugel en femurhals. Op basis van deze bevindingen onderging de patiënt in Juli en September 2009 een operatie bestaande uit curettage en opvulling met zowel een autologe graft als botvervangers van het derde linkerhandhandsbeentje en de linker supra-acetabulaire laesie. Het pathologische laboratorium meldde een GCT. Latere controles met CT en MRI van de borst en het bekken toonden de vergroting van polylobulated en expansieve lytische laesies in het bekken (Fig. 1), heiligbeen, rechter femurhals en L5, met het verschijnen van een nieuwe laesie in de rechter femurkop en linker zevende costale boog (Fig. 2). Deze veranderingen werden toegeschreven aan gemetastaseerde tumorprogressie. Vanwege aanhoudende pijn die de ambulatie volledig verhinderde, werd de patiënt doorverwezen naar de afdeling bottumor van Hospital Universitario La Paz, waar een overzicht van pathologische monsters leidde tot de conclusie dat de botlaesies zeer suggestief waren van reuze cel reparatieve granulomen en histologisch niet te onderscheiden van bruine tumoren. Hyperparathyreoïdie werd daarom uitgesloten. In November 2010 werd de patiënt doorverwezen naar de afdeling endocrinologie, waar aanvullende laboratoriumtests de volgende resultaten opleverden: totaal calcium 14mg/dL, gecorrigeerd calcium 13,2 mg/dL, ionisch calcium 1,72 mmol/l, fosfaat 1,9 mg/dL, magnesium 1,86 mg/dL, urinair calcium 968,60 mg/24h, creatinine 0,55 mg/dL, iPTH 535pg/mL, vitamine D 13ng/mL. Totale lichaam CT toonde een knobbeltje 1,5 cm in diameter in de theoretische locatie van de rechter bijschildklier, en een bijschildklier scan met 20mCi van TC 99-sestamibi onthulde bevindingen consistent met rechts hyperfunctionerend bijschildklier adenoom. Bovendien werd de aanwezigheid van een geassocieerd feochromocytoom uitgesloten gebaseerd op zowel biochemie als morfologie. Rechter parathyroïdectomie werd uitgevoerd, en een intraoperatieve biopsie werd gemeld als een parathyroïd adenoom. Het Laatste histopathologische onderzoek bevestigde de aanwezigheid van een bijschildklieradenoom van 4,5 g in gewicht en 2,2 cm×2 cm×1,9 cm in grootte.
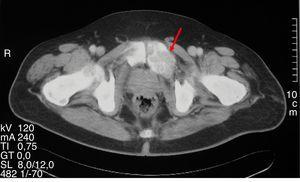
figuur 1.
expansieve lytische laesie overeenkomend met osteïtis fibrosa cystica in een bekken CT-scan.
(0.14 MB).
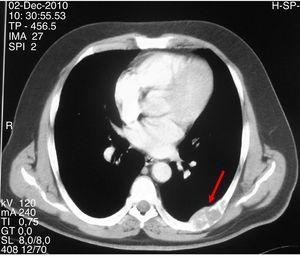
Figuur 2.
CT-scan toont een expansieve lytische laesie met lobulaire contouren en corticale verdunning in de zevende linker costale boog.
(0.21 MB).
na de operatie ervoer de patiënt symptomatische hypocalciëmie die behandeling met calcitriol en calcium vereiste, die tot op heden is voortgezet. Hij wordt nog steeds gevolgd op de afdeling endocrinologie, rapporteert significante symptomatische verbetering en is in staat om te lopen met krukken. Een genetische studie vond geen mutaties in het Men-1 gen.
GCT is een sterk vasculariseerde tumor die wordt gevonden in de metafysen of epifysen van lange botten of in het bekken, heiligbeen of wervels.2 de radiografische en histologische verschijning van bruine tumoren typisch voor OFC kan nauw na te bootsen een GCT, zoals opgetreden in onze patiënt, en differentiatie moet worden gemaakt op basis van klinische symptomen en laboratoriumresultaten (iPTH). Sommige van de auteurs3, 4 hebben gevallen van OFC gemeld waarbij secundaire metastatische botziekte aanvankelijk werd vermoed op basis van klinische symptomen en radiografische beelden. Bij onze patiënt was de diagnose van een gemetastaseerde primaire bottumor echter gebaseerd op histologische bevindingen, terwijl de familiegeschiedenis, die in de bovenstaande gevallen niet werd gerapporteerd, consistent was met PHP.
aan de andere kant wordt vitamine D-deficiëntie vaak gedetecteerd bij patiënten met PHP en is3, 5 geassocieerd met exacerbatie van de biochemische en fenotypische presentatie van de ziekte (hogere serum PTH-spiegels,grote bijschildklieradenomen, en een groter risico op fracturen), wat kan hebben bijgedragen aan het Floride klinische beeld van onze patiënt.Het is bekend dat familiaire vormen van hyperparathyreoïdie soms voorkomen (5%), en de meest voorkomende oorzaken zijn multipele endocriene neoplasie (mannen) type 1-en 2A-syndromen, hyperparathyreoïdie-kaak tumor (HPT-JT) syndroom en familiaire geïsoleerde hyperparathyreoïdie (FIHP).Bij mannen 1 is hyperparathyreoïdie de vroegste en meest voorkomende presentatie (>90%), terwijl deze bij mannen 2A laat voorkomt en een lage penetrantie heeft. Hoewel de genetische studie voor mannen 1 negatief was bij onze patiënt, moet worden opgemerkt dat een vals-negatief resultaat kan optreden in maximaal 30% van de geteste gevallen als gevolg van mutatiepatronen waarbij verschillende gengebieden betrokken zijn of mutaties in tot nu toe onbekende genen die de transcriptie of werking van mannen beïnvloeden.7 Dit, samen met het waarschijnlijke asynchrone optreden van verschillende aspecten van MEN-1, maakt continue monitoring noodzakelijk. Men 2A is onwaarschijnlijk in afwezigheid van Schildklier neoplastische betrokkenheid of feochromocytoom. Differentiële diagnose moet ook HPT-JT omvatten vanwege de schaal van botbetrokkenheid en de grote omvang van het adenoom. De definitieve bevinding van een bijschildkliercarcinoom zou deze diagnose hebben ondersteund, vanwege zijn frequente voorkomen in HPT-JT.Echter, de afwezigheid van mandibulaire of maxillaire fibro-osseuze laesies en renale laesies maakte dit onwaarschijnlijk. Ten slotte kan, hoewel FIHP in sommige gevallen een variant van andere hyperparathyroïdsyndromen kan vertegenwoordigen, de mogelijkheid niet worden uitgesloten dat mutaties die zich in nog niet geïdentificeerde loci bevinden, anders dan die welke bij mannen 1 en 2 en bij HPT-JT zijn gemeld, dit syndroom kunnen veroorzaken.
het belang van het gerapporteerde geval ligt in het feit dat het het belang illustreert van het beoordelen van het fosfor-en calciummetabolisme en de bijschildklierfunctie bij alle patiënten met botlaesies, van het vermoeden van een potentieel PHP als suggestieve laesies bestaan, en van het zoeken naar een waarschijnlijke onderliggende genetische component door middel van een gedetailleerde familie-en persoonlijke geschiedenis.