Wij verzamelden vruchten van barbarum L. en L. ruthenicum op vijf ontwikkelingsstadia, van jong fruit (≈10 dagen na de bloei) om te rijpen (rijp) fruit (34-45 dagen na de bloei), en bestudeerde hun transcriptoom en metaboloom.
RNA-seq de novo assemblage en functionele annotatie van unigenen
We bereidden in totaal 30 cDNA-bibliotheken van vruchten van L. barbarum en L. ruthenicum, met drie biologische replicaten (drie vruchten van drie bomen) op elk tijdstip: 2 soorten × 5 tijdpunten × 3 biologische replicaten. Monsters werden geëtiketteerd LB/LR (1-5)-(1-3), waarbij LB L. barbarum is en LR L. ruthenicum, 1-5 zijn ontwikkelingsstadia van fruit (S1-S5), en 1-3 zijn individuele monsters( biologische replicaten); dus bijvoorbeeld LB1-1 staat voor L. barbarum, 1e bemonsterde ontwikkelingsfase (S1), vruchtmonster nr. 1 (van de drie). We genereerden meer dan 1,72 miljard pair-end reads voor deze 30 cDNA-bibliotheken, wat overeenkomt met een gemiddelde van 57,2 miljoen reads per sample (aanvullende Dataset S1). Strenge kwaliteitsbeoordeling en gegevensfiltering leverden in totaal 801.766 hoogwaardige reads op met een gemiddelde lengte van 730 en N50 van 1107 bp (Tabel 1). Uiteindelijk werden in totaal 326.276 unigenes met een gemiddelde lengte van 596 bp en N50 van 847 bp verkregen uit de transcripten (Tabel 1). Correlatiecoëfficiënten voor RNA-seq gegevens voor de 30 steekproeven wijzen op zeer goede consistentie van resultaten onder biologische replicaten (Fig. 2).
Tabel 1 Kenmerken van geassembleerde transcripten en unigenes.
Figuur 2
Heatmap van correlatiecoëfficiënten voor RNA-seq gegevens voor 30 monsters van L. barbarum (LB) en L. ruthenicum (LR) vruchten in vijf verschillende ontwikkelingsstadia. De monsters zijn voorzien van het etiket LB / R1-5_1-3, waarbij LB L. barbarum is, LR L. ruthenicum, 1-5 zijn vrucht ontwikkelingsstadia, en 1-3 individuele monsters. Monsters werden gegroepeerd door hiërarchische clustering; dendrograms boven en links van de heatmap wijzen op verwantschap van monsters.
van alle 326.276 unigenen die werden opgevraagd tegen openbare databases, werden in totaal 193.021 (59,15%) overeenkomende genen en/of eiwitten in ten minste één database, en 12.171 (3,73%) werden geannoteerd in alle databases. Het grootste aantal unigenes (149,863, 45,93%) werd geannoteerd in de nt-database, en het laagste aantal (24,017; 7,36%) in de KOG-database.
de hoogste aantallen DEGs werden geïdentificeerd in alle paarsgewijze vergelijkingen van de 1ste fase, en in de 2de vs.5de fase vergelijking (alle > 10.000 degs; Tabel 2). De kleinste aantallen werden geïdentificeerd in vergelijkingen van de 3e vs. 4e en 4e vs. 5e fase (255-257). Heatmap analyse van DEGs in LB toont aan dat vrij verschillende sets van genen zeer upregulated waren in de vroege ontwikkelingsstadia (1 + 2) en in latere stadia (3 tot 5) (Fig. 3A). Uit de analyse van de verwantschap van het monster blijkt dat de monsters in twee klassen kunnen worden verdeeld (fasen 1 + 2 en 3 + 4 + 5), met de laatste clade verder onderverdeeld in twee clades: fase 3 + 4 en Fase 5. Intraspecifieke Kegg functionele classificatie analyse van deze DEGs identificeerde 15 routes significant (p < 0,05) differentieel gereguleerd tussen verschillende ontwikkelingsstadia (aanvullende Dataset S2). Bijzonder sterk differentieel gereguleerd waren ‘signaaltransductie van plantenhormonen’, ‘ biosynthese van fenylpropanoiden (b.)’, ‘linolzuurmetabolisme (m.)’, ‘ zetmeel en sucrose m.”, en “zeatin b.” (Fig. 3A).
Tabel 2 totale aantallen significant gereguleerde genen en metabolieten in paarsgewijze vergelijkingen van ontwikkelingsstadia bij de twee onderzochte species.
Figuur 3
Heatmaps en functionele pathway analyses van differentieel tot expressie gebrachte genen (degs) in Lycium barbarum (panel A) en L. ruthenicum (panel B) vruchten. Heatmaps werden gegenereerd door een hiërarchische analyse van DEGs (y-as) en individuele monsters (x-as), waarbij dendrograms boven en links van de heatmap wijzen op verwantschap van monsters. De monsters worden geëtiketteerd op LB / R_1-5_1-3, waarbij het acroniem voor de soort (LB of LR) wordt gevolgd door het ontwikkelingsstadium van de vrucht (1-5) en het nummer van het monster (1-3). Intraspecifieke Kegg-pathway-analyses van DEGs in alle vijf ontwikkelingsstadia van de twee soorten worden rechts van de heatmaps getoond. Alleen de top 15 verrijkte paden worden vermeld. q-waarde is een voor FDR gecorrigeerde p-waarde.
voor een meer diepgaande analyse van de gegevens, hebben we ons gericht op de vergelijking van de meest significant gereguleerde routes in opeenvolgende ontwikkelingsstadia. In de eerste paarsgewijze vergelijking (S1 versus S2) was ‘phenylpropanoid b.’ de hoogst differentieel gereguleerde route, gevolgd door ‘zetmeel en sucrose m.’ (Fig. 4). In beide routes werden zeer grote aantallen DEGs (>100) geïdentificeerd. Een vergelijkbaar resultaat werd waargenomen in de volgende paarsgewijze vergelijking, S2 vs. S3, maar ondanks het vrij grote aantal DEGs (>80) ‘zetmeel en sucrose m.’vertoonde iets lagere q-waarde. In de S3 vs. S4 vergelijking was ‘carbon fixation in photosynthetic, organisms’ de meest significante gereguleerde route, maar het aantal genen was veel lager. In het laatste paar, S4 vs. S5, ‘zeatine b.’, flavonoïde b.’, vetzuur b.’ en ‘galactose m.’ waren de meest significant gereguleerde routes, maar geen van de routes vertoonde meer dan twee DEGs.
Figuur 4
lb transcriptome: paarsgewijs interstage KEGG metabolische route verrijking. (A) Fase 1 vs.fase 2. (B) fase 2 vs. fase 3. (C) Fase 3 vs.fase 4. (D) fase 4 vs.fase 5. Top 15 (of alle als totaal <15) aanzienlijk verrijkte routes worden getoond. Kleur van de balk staat voor de grootte van de q-waarde (een voor FDR gecorrigeerde p-waarde). Kleurenkaart is weergegeven in de figuur.
lb Metabolome
Wij voerden paarsgewijze ontwikkelingsstadiumvergelijkingen uit om de verrijking van metabolites tussen alle stadiaparen te identificeren. Het aantal differentieel gereguleerde metabolieten in paarsgewijze vergelijkingen van ontwikkelingsstadia in LB varieerde van 66 (stadia 3 vs.4) tot 129 (stadia 2 vs. 5). Het grootste aantal in opeenvolgende etappevergelijkingen werd waargenomen tussen de eerste en de tweede etappe (117). Deze werden toegewezen aan een groot aantal routes; met het grootste aantal metabolieten toegewezen aan ‘ B. van secundaire metabolieten ‘(ook de laagste p-waarde), gevolgd door ‘eiwit spijsvertering en absorptie’ en ‘B. van aminozuren’ (Fig. 5; Aanvullende Cijfers). In de opeenvolgende fase paar vergelijking (S2 vs. S3),’ ABC transporters ‘was de meest significant verrijkte route, gevolgd door’purine m.’. In de vergelijking van S3 versus S4-paren was ‘microbiële m. in diverse omgevingen ‘de meest significant verrijkte route, gevolgd door’carbapenem m.’. In de vergelijking van het S4-Versus S5-paar vertoonde een relatief groot aantal routes vergelijkbare resultaten (twee metabolieten en vergelijkbare p-waarden), maar opmerkelijk is het verschijnen van ‘isoflavonoid b.’ en ‘flavonoid b.’ onder hen (Fig. 5; Aanvullende Cijfers).
Figuur 5
lb metabolomic gegevens: paarsgewijs interstage KEGG metabolische route verrijking. (A) Fase 1 vs.fase 2. (B) fase 2 vs.fase 3. (C) Fase 3 vs.fase 4. (D) fase 4 vs.fase 5. Rijke factor is de verhouding tussen het aantal significant gereguleerde metabolieten in de route en het totale aantal metabolieten dat in die route is geannoteerd (bereik = 0 tot 1,0). De grootte van het punt staat voor het aantal aanzienlijk verrijkte metabolieten in de overeenkomstige route, en de kleur van het punt staat voor de P-waarde (beide legendes in de figuur).
wat de individuele metabolieten betreft, werd bij de vergelijking van S1 vs.S2 de lijst van metabolieten die in S1 upreguleerd werden aangevuld met Trehalose, Glactinol en L-appelzuur (alle ≈20 log2FC). Oliezuur, 2-Oxoadipinezuur en stearinezuur waren de hoogst gereguleerde metabolieten in S2 (alle ≈15 tot 18 log2FC). De lijst van meest upregulated metabolieten in S2 in vergelijking met S3 werd aangevuld met dihydroxyaceton, LysoPC(18:1(9Z)) en Adenine (alle ≈16 tot 17,5 log2FC). De lijst van de meest upregulerende metabolieten in S3 (vergeleken met S2) werd aangevuld met Trehalose, Galactinol en L-appelzuur (alle ≈19 tot 20 log2FC). In de S3 vs. S4 vergelijking werd de lijst van de meest upregulerende metabolieten in de S3 aangevuld met L-appelzuur, DL-Arginine en oliezuur (alle ≈18 tot 19 log2FC), terwijl de lijst van de meest upregulerende metabolieten in de S4 werd aangevuld met D-Mannose, n-Acetyllactosamine en LysoPC(18:1(9Z)) in LB (alle ≈16 log2FC). In de S4 vs. S5 vergelijking, de lijst van de meest upregulated metabolieten in de S4 werd aangevuld met Trehalose, palmitinezuur, n-Acetyllactosamine (alle ≈17 tot 18 log2FC) in LB. De lijst van de meest upregulerende metabolieten in de S5 werd aangevuld met L-Norleucine, Antranilzuur (vitamine L1) en DL-Arginine (alle ≈16 tot 18 log2FC) (aanvullende Dataset S3).
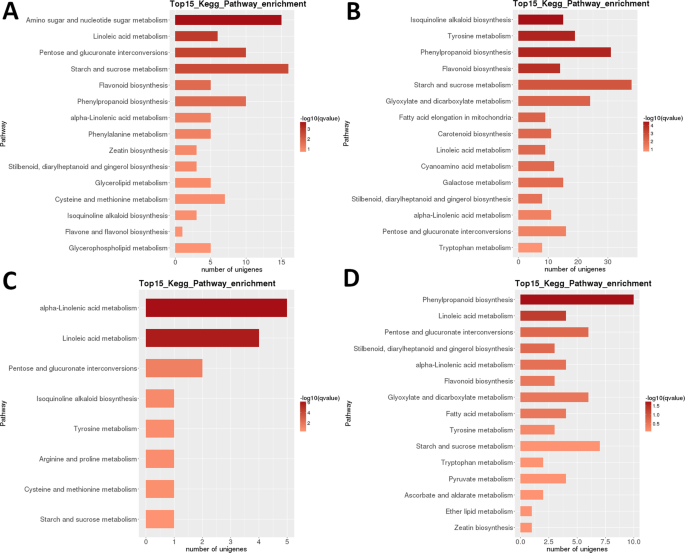
Lycium ruthenicum (LR): uit de analyse van de Heatmap van DEGs in LR blijkt dat bijna volledig verschillende genensets in de vroege ontwikkelingsstadia (1 + 2) en in rijpe vruchten (Stadium 5) sterk werden opgewaardeerd, met een duidelijke transcriptomische reset na de tweede fase (Fig. 3B). Uit de analyse van de verwantschap van het monster blijkt dat de monsters in twee klassen kunnen worden verdeeld (fasen 1 + 2 en 3 + 4 + 5), met de laatste clade verder onderverdeeld in twee clades: fase 3 + 4 en Fase 5. Intraspecifieke Kegg functionele classificatie analyse van deze DEGs identificeerde 35 routes significant (p < 0,05) differentieel gereguleerd tussen verschillende ontwikkelingsstadia (aanvullende Dataset S2). Bijzonder sterk differentieel gereguleerd waren ‘fotosynthese’, ‘ linolzuur m.’, ‘ isochinoline alkaloïde b.’, ‘flavonoïde b.’ en ‘zetmeel en sucrose m’ (Fig. 3B). De hoogste aantallen DEGs werden geïdentificeerd in de meeste paarsgewijze vergelijkingen van Fase 1 (1 vs.2 was een uitzondering), en in de vergelijking van Fase 2 vs. 5 (Alle >5.000 DEGs; Tabel 2). Veruit het kleinste aantal werd geïdentificeerd in de S3 vs.S4 vergelijking (39). In de paarsgewijze vergelijking van S1 versus S2 was ‘aminosuiker en nucleotidesuiker m. ‘de hoogst differentieel gereguleerde route, gevolgd door’ linolzuur m. ‘ (Fig. 6). In de paarsgewijze vergelijking van S2 versus S3 waren de meest significant differentieel gereguleerde routes ‘isochinoline-alkaloïde b.’, ‘ tyrosine m.’, ‘fenylpropanoide b’en’ flavonoïde b.’. In beide paarsgewijze vergelijkingen werd het hoogste aantal DEGs (respectievelijk>15 en >35) vastgesteld in de ” zetmeel-en sacharose-m.”. In de S3 vs. S4 vergelijking,’ (alfa-)linolzuur m. ‘ was de meest significant gereguleerde route, maar het aantal genen was veel lager. In de S4 vs. S5 vergelijking waren ‘phenylpropanoid b’ (ook het grootste aantal DEGs) en ‘linolzuur m.’ de meest significant gereguleerde routes.
Figuur 6
LR transcriptome: paarsgewijs interstage Kegg metabolische route verrijking. (A) Fase 1 vs.fase 2. (B) fase 2 vs.fase 3. (C) Fase 3 vs.fase 4. (D) fase 4 vs.fase 5. Top 15 (of alle als totaal <15) aanzienlijk verrijkte routes worden getoond. Kleur van de balk vertegenwoordigt de grootte van de q-waarde (een FDR-aangepaste p-waarde, kleurenkaart weergegeven in de figuur).
LR Metaboloom
het aantal differentieel gereguleerde metabolieten in paarsgewijze vergelijkingen van ontwikkelingsstadia in LR varieerde van 66 (stadia 3 vs.4) tot 133 (stadia 2 vs. 5) (aanvullende Dataset S3). In opeenvolgende etappevergelijkingen werd het grootste aantal waargenomen tussen de eerste en de tweede etappe (117). Deze werden toegewezen aan een groot aantal trajecten; met het grootste aantal metabolieten dat is toegewezen aan “B van secundaire metabolieten”, gevolgd door “eiwitvertering en-absorptie”, “B van aminozuren” en “flavonoïde b”. In de S2 vs. S3 vergelijking,’ ABC transporters ‘was de meest significant verrijkte route, gevolgd door’purine m’. In de vergelijking van S3 versus S4-paren was ‘microbiële m. in diverse omgevingen ‘de meest significant verrijkte route, gevolgd door’carbapenem m.’. In de S4 vs. S5 paar vergelijking, een relatief groot aantal routes vertoonde vergelijkbare resultaten (2 metabolieten en soortgelijke p-waarden), maar opmerkelijk is het verschijnen van ‘isoflavonoid b.’ en ‘flavonoid b.’ onder hen (Fig. 7; Aanvullende Cijfers).
Figuur 7
de gegevens van LR metabolomic: paarsgewijs INTERSTAGEKEGG metabolische wegverrijking. (A) Fase 1 vs.fase 2. (B) fase 2 vs.fase 3. (C) Fase 3 vs.fase 4. (D) fase 4 vs.fase 5. Rijke factor is de verhouding tussen het aantal significant gereguleerde metabolieten in de route en het totale aantal metabolieten dat in die route is geannoteerd (bereik = 0 tot 1,0). De grootte van het punt staat voor het aantal aanzienlijk verrijkte metabolieten in de overeenkomstige route, en de kleur van het punt staat voor de P-waarde (beide legendes in de figuur).
wat betreft individuele metabolieten (aanvullende gegevensset S3), in de S1 vs. S2 vergelijking, de lijst van metabolieten upregulated in de S1 werd aangevuld met Trehalose, Galactinol en L-appelzuur (≈19-21 log2FC), terwijl oliezuur, 2-Oxoadipinezuur en stearinezuur waren de meest upregulated metabolieten in S2 (≈15-18 log2FC). In de S2 vs.S3 vergelijking waren dihydroxyaceton, Indoxylsulfaat en n-Acetyllactosamine de meest upregulerende metabolieten in de S2 (≈17-19, 5 log2FC), en Trehalose, Galactinol en L-appelzuur (≈19-21 log2FC) in de S3. In de S3 vs. S4 vergelijking, l-appelzuur, DL-Arginine en oliezuur werden upregulated in de S3 (≈16-19 log2FC), en 1,7-Dimethylxanthine, D-Mannose en N-Acetyllactosamine (≈15-17 log2FC) in de S4. In de S4 vs.S5 vergelijking werden Flavinmononucleotide, Trehalose en Isoferulaatzuur verhoogd in de S4 (≈18-20 log2FC), terwijl PG(16:0/18:1(9Z)), D-Proline en DL-Arginine het hoogst verhoogd waren (alle ≈16 tot 18 log2FC) metabolieten.
interspecifieke vergelijkende analyse van DEGs in verschillende ontwikkelingsstadia
totaal DEGs tijdens de fruitontwikkeling
vergelijking van de interspecifieke paarsgewijze fase (LR1 vs. LB1, LR2 vs. LB2, enz.) toont aan dat 928 DEGs werden gedeeld door alle vijf paren (Fig. 8A). Het hoogste aantal DEGs werd vastgesteld in Fase 3 (3989), en het laagste in Fase 4 (2825) (Fig. 8B); terwijl de hoogste aantallen degs die uniek zijn voor een paar werden waargenomen in fasen 3 (574), 1 en 5 (beide 554), en de laagste in Fase 4 (126) (Fig. 8A). De aantallen op – en neergereguleerde DEGs waren in elk van de paarsgewijze etappevergelijkingen relatief gelijk; in Fase 5 werden bijvoorbeeld 1668 DEGs verhoogd en 1670 DEGs verlaagd in LR in vergelijking met LB (Fig. 8 ter). Echter, in de andere vier stadia was het aantal upregulated genen lichtjes (93 tot 189 DEGs) hoger.
Figuur 8
genen die differentieel tot expressie komen (degs) tussen vruchten van L. barbarum (LB) en L. ruthenicum (LR). (A) gedetailleerde stagegewijze (1 tot 5) vergelijkingen (LB vs.LR). (B) het aantal opregulerende (rode) en opregulerende (groene) DEGs in LR in vergelijking met LB in vijf bestudeerde ontwikkelingsstadia.
Transcriptome-pathways
Heatmap analyse van Degs geeft aan dat de vruchten van twee soorten zeer verschillende genexpressieprofielen vertonen gedurende alle ontwikkelingsstadia, maar biologische replicaten vertonen zeer vergelijkbare profielen, wat wijst op een beperkte mate van individuele variabiliteit in elk ontwikkelingsstadium (aanvullende cijfers: Fig. S9). Vergelijkende analyse van Kegg-pathway-verrijking toont aan dat slechts enkele routes consistent sterk verrijkt waren (in termen van genregulatie) in LR in vergelijking met LB gedurende alle vijf ontwikkelingsstadia (Fig. 9). Met name de plant hormoon signaaltransductie (2de hoogste in S1, 8-hoogste in S2, de hoogste in de S3, S4 en S5) en plant-pathogeen interactie (de hoogste in S1, 3e hoogste in S2, 2de hoogste in S3, 4e hoogste in S4, en de 15e-hoogste in S5) waren relatief hoog up gereguleerd in alle stadia. De biosynthese van Phenylpropanoid (niet in top 15 in S1, hoogste in s2, 3de-hoogste in S3, 7de-hoogste in S4, 11de-hoogste in S5), ubiquinon en andere biosynthese van terpenoid-quinone (niet in top 15 in S1, 6de-hoogste in s2, 7de-hoogste in S3, 2de-hoogste in S4, 6de-hoogste in S5) waren ook relatief hoogregulerend in alle stadia behalve de eerste. Flavonoïde biosynthese route was niet sterk verrijkt in vroege stadia (niet in top 15 in S1, 14e-hoogste in S2), en sterk verrijkt in late stadia (3e tot 4e-hoogste in stadia 3 tot 5). (Alfa-)Linolzuurmetabolisme was sterk verrijkt in de middelste stadia (9e-hoogste in S1, 2e-hoogste in S2, 5e en 6e-hoogste in S3, 8e-hoogste in S4, niet in top 15 in S5).
figuur 9
vergelijkende analyse van de verrijking van de KEGG-metabole route. De top 15 paden verrijkt in L. ruthenicum in vergelijking met L. barbarum zijn weergegeven aan de linkerkant (rood), en degenen verrijkt in L. barbarum in vergelijking met L. ruthenicum aan de rechterkant (groen). Ontwikkelingsstadia (1-5) worden aangegeven in de figuur. q-waarde is een voor FDR gecorrigeerde p-waarde.
onder de paden die in L. ruthenicum zijn neergestort in vergelijking met L. barbarum (Fig. 9) Er werden opmerkelijke veranderingen waargenomen tussen de vroege stadia (1 en 2), toen het cyanoaminozuurmetabolisme en de biosynthese van carotenoïden het sterkst gedegradeerd waren, en de late stadia (4 en 5), toen de SNARE-interacties in vesiculair transport, het nicotinaat-en nicotinamidemetabolisme en het porfyrine-en chlorofylmetabolisme constant relatief sterk gedegradeerd waren.
Transcriptoom – individuele genen
sommige van de genen die het meest differentieel tot expressie kwamen, waren specifiek voor ontwikkelingsstadia (d.w.z. sterk differentieel gereguleerd alleen in vroege of late ontwikkelingsstadia), maar sommige waren consistent sterk differentieel gereguleerd in alle vijf onderzochte stadia (aanvullende Dataset S4). Verscheidene immuniteits-verwante genen zeer hoogst upregulated in LR in vergelijking met LB in de vroege ontwikkelingsstadia zijn onder de voorbeelden van het ontwikkelingsstadiumspecifieke uitdrukkingspatroon: chitinase was de 2de hoogste upregulated DEG in S1 (13,43-voudig), de hoogste in S2 (13,89-voudig), maar in latere stadia was het geen DEG. Op dezelfde manier, EIX receptor 1/2 was ook zeer hoogst upregulated in vroege stadia, hoogste in S1 (13.70) en tweede-hoogste in s2 (10.90), maar het werd ook niet geà dentificeerd als DEG in latere stadia. Sommige groei-gerelateerde genen vertoonden ook een soortgelijk expressiepatroon: fosfoglyceraatkinase (PGK) was een van de handvol hoogst upregulated genen in de eerste drie stadia (13.14, 12.87 en 12.77 respectievelijk), maar het werd ook niet geïdentificeerd als DEG in latere stadia. CCR4-niet transcriptie complexe subeenheid 7/8 (CNOT7/8) vertoonde ook een zeer vergelijkbaar expressiepatroon: zeer upregulated in de eerste drie fasen, en niet een DEG in de fasen 4 en 5. Verscheidene flavonoid en phenylpropanoid biosynthese-geassocieerde genen vertoonden een omgekeerd ontwikkelingsstadium-specifiek uitdrukkingspatroon, met vrij lage uitdrukking in vroege stadia, en zeer hoog in latere stadia. Voorbeelden hiervan zijn: bifunctionele dihydroflavonol 4-reductase/flavanon 4-reductase (DFR), die in S1 (2) lichtjes werd verhoogd in LR.25), Geen graad in S2, hoogregulerend in S3 (7.79), en derde hoogstregulerend gen in S4 (14.25) en S5 (16.03). Een paralogue van dit gen vertoonde een bijna identiek patroon: lichtjes opgereguleerd in s1 (2,44), geen DEG in S2, hoog opgereguleerd in S3 (7,40), 6e-hoogst opgereguleerd DEG in S4 (13,26) en 5e in S5 (14,59). Op dezelfde manier was flavonoïde 3′,5′-hydroxylase (F3’5′ H) geen DEG in de eerste twee stadia, hoogregulerend in S3 (6.69), 5de hoogstregulerend gen in S4 (13.42), en 4de hoogst in S5 (15.05). Flavonoïde o-methyltransferase (OMT) was geen DEG in de S1, maar in S2 vertoonde het al een middelhoge upregulatie (4.32), door de S3 was het al de derde hoogste upregulated DEG (13.30), en het was het hoogst-upregulated gen in S4 (18.73) en S5 (18.10). Leucoanthocyanidin dioxygenase (LDOX; biosynthese van anthocyanine) was geen DEG in S1 en S2, gevolgd door hoge tot zeer hoge upregulatie in latere stadia (respectievelijk 5,63, 9,44, 11.56). Twee chalconesynthase-paralogen (CHS en CHS2; flavonoïde biosynthese) werden ook niet sterk gereguleerd in S1 en S2 (CHS2: geen DEG, CHS: -1.14 in S1, geen DEG in S2), maar in S3–S5 vertoonden beide genen een middelhoge tot hoge upregulatie (respectievelijk CHS2: 5.32, 7.84, 6.00; en CHS: 4.67, 7.01, 6,82;). We selecteerden deze genen voor qPCR analyse, en de resultaten zijn zeer congruent met de RNA-seq gegevens (aanvullende resultaten; aanvullende Dataset S5). Ten slotte werd cytokininedehydrogenase, een zeatine-biosynthese-gerelateerd gen, ook in toenemende mate upregulated tijdens de laatste drie stadia (2.6-5.7).
sommige genen werden echter consistent differentieel tot expressie gebracht in alle vijf onderzochte stadia. Voorbeelden ook een aantal immuniteit-gerelateerde genen, zoals twee paralogues van glutathion-S-transferase, zeer up gereguleerd in LR in vergelijking met LB in alle fasen: 9.38 en 8,58 (alle waarden worden gepresenteerd als fold-wijzigingen in respectievelijke volgorde) in S1, 6.30 en 6.34 in S2, de 2e en 7e hoogste up gereguleerd Gr in S3 (14.08 en 12.70), 2e en 4e hoogste in S4 (15.71 en 14.16) en 2e en 6e hoogste in S5 (16.48 en 14.40). Plant disease resistance protein RPM1 was ook sterk upregulated in alle vijf stadia (S1 = 13,15; S2 = 12,08; S3 = 13,11, S4 = 12,81; S5 = 13:94). Onder de consequent differentially uitgedrukte genen door alle ontwikkelingsstadia waren ook wat gerelateerd aan het aminozuurmetabolisme, maar hun patroon werd omgekeerd in vergelijking met vorige voorbeelden: zij tentoongesteld hoge downregulation in LR in vergelijking met LB. Voorbeelden zijn acetyl-CoA acyltransferase 1 (AAT1; valine, leucine en isoleucine afbraak), met een temporaal profiel van steeds hogere downregulatie, beginnend van -7,0 in de S1 tot <−10-voudig in de laatste drie stadia. Proline iminopeptidase, geassocieerd met arginine en proline metabolisme, werd in alle stadia sterk verlaagd in LR: S1 = -9,75, S2 = -10,89 (3e hoogste), S3 = -11,05 (4e hoogste), S4 = -10,01, en S5 = -11,98 (3e hoogste). Ten slotte werd 5–methyltetrahydropteroyltriglutamaat-homocysteïne methyltransferase (metE) consequent extreem sterk verlaagd in LR in alle stadia: 2e hoogste in S1 (-11,76), de hoogste in S2 (-11,74), 3e hoogste in S3 (-11,43), de hoogste in S4 (-12,36), en 2e hoogste in s5 (-12,83). Twee DNA-replicatie en transcriptie-geassocieerde genen werden ook sterk downregulated in LR in alle stadia: GTP-bindende kernproteã ne liep (liep; -10.0 aan -12.0) en replicatiefactor A1 (RFA1) (-8.0 aan -12.0). Sommige groei-en stressgerelateerde genen werden ook consequent sterk gedegreguleerd in LR: heterogeen kernribonucleoproteïne A1/A3 (Hnrnp; -7 tot -11) en hitteschok 70 kDa eiwit 1/8 (HSPA1_8) S1 = -4,95, S2 = -8,88, S3 = -11,48 (tweede hoogste), S4 = -9,36, S5 = -12,89 (de hoogste). Intrigerend was een phenylpropanoid biosynthese-gerelateerd gen, shikimate hydroxycinnamoyltransferase (HCT), ook consistent sterk gedegradeerd in LR: S1 = -6.82, S2 = -8,14, S3 = -11,71 (de hoogste), S4 = -11,00 (derde hoogste), S5 = -11,91 (vierde hoogste). Echter, een belangrijke regulator van anthocyanine biosynthese, transcriptiefactor MYB114, was zeer upregulated in LR tijdens alle vijf ontwikkelingsstadia: 6.11, 4.69,7.47, 9.05, en 8.95 (respectievelijk S1–S5).
Metaboloom-routes
we hebben ook een vergelijkende interspecifieke fase-wise analyse van metabole routes uitgevoerd (Fig. 10). In de eerste ontwikkelingsfase (S1) identificeerden we 39 differentieel gereguleerde metabolieten. Onder de top 20 wegen werden deze metabolites geassocieerd met, verscheidene van hen werden geassocieerd met aminozuren, maar het totale aantal metabolites per weg waren vrij klein (1-2), en P-waarden suggereerden geen hoog niveau van significantie (Fig. 10-paneel 1). Met name het metabolisme van vitamine B6 (m) en microbiële m in diverse milieu ‘ s vertoonden beiden relatief hoge P-waarden, verrijkingsfactor (EF) van 1,0, en 2 geïdentificeerde metabolites. In de S2 identificeerden we 58 differentieel gereguleerde metabolieten, geassocieerd met slechts vier routes: tryptofaan m, fenylpropanoide biosynthese (b), B van fenylpropanoiden (dit zijn twee verschillende routes in de Kegg-database), en fenylalanine, tyrosine en tryptofaan b (alle EF = 1,0, 2-3 metabolieten, en p < 0,5; Fig. 10-paneel 2). In de S3 identificeerden we 59 differentieel gereguleerde metabolieten, geassocieerd met 19 routes, meestal allemaal met de EF 1.0, maar relatief niet-significante P-waarden (>0,5; Fig. 10-panel 3). Routes met een relatief hoog aantal metabolieten (n = 5) waren: eiwitvertering en-absorptie, B van secundaire metabolieten van planten, B van antibiotica en B van aminozuren. In de S4 identificeerden we 58 differentieel gereguleerde metabolieten, geassocieerd met een groot aantal routes, meestal met de EF 1.0, en relatief hoge significantiewaarden (meestal P > 0,5; Fig. 10-paneel 4). Routes met een relatief hoog aantal metabolieten (n ≥ 3) waren: fenylpropanoide b, fenylalanine, tyrosine en tryptofaan b, glucosinaat b, B van alkaloïden afgeleid van shikimaat route, en 2-oxocarbyl m. in de rijpe vrucht (S5), identificeerden we 39 differentieel gereguleerde metabolieten, geassocieerd met een groot aantal routes, maar meestal met lage P-waarden en slechts 1 metaboliet per route (Fig. 10-paneel 5). Routes met meer dan 1 metaboliet werden: eiwit spijsvertering en absorptie, phenylpropanoid b, minerale absorptie, centrale koolstof m in kanker, B van secundaire metabolieten, B van phenylpropanoids, en aminoacy tRNA B. de belangrijkste component analyse (PCA) van alle gegevens (2 species × 5 stadia × 5 biologische replicaten) onthulde hoge gelijkenis tussen biologische replicaten (clustering), en bevestigde opmerkelijke variabiliteit tussen verschillende vrucht rijping stadia voor beide species (Fig. 10-paneel 6).
Figuur 10
vergelijkende analyse van de verrijking van de KEGG-metabole route. De top 15 paden verrijkt in L. ruthenicum in vergelijking met L. barbarum zijn weergegeven aan de linkerkant (rood), en degenen verrijkt in L. barbarum in vergelijking met L. ruthenicum aan de rechterkant (groen). Ontwikkelingsstadia (1-5) worden aangegeven in de figuur. q-waarde is een voor FDR gecorrigeerde p-waarde.
Metabolome-individuele metabolites
de lijst (aanvullende Dataset S6) van hoogst differentieel geregelde metabolites tussen de twee species, vertoonde enige variatie tussen de vijf stadia van de fruitontwikkeling. Intrigerend, Fructose 1-fosfaat was de meest upregulated metaboliet in LR, vergeleken met LB, gedurende alle vijf stadia: log2 vouw verandering = 6.3, 7.6, 7.7, 8.1, en 6.5 (stadia 1 tot 5 respectievelijk). Met betrekking tot de metabolieten die in LB hoog zijn gereguleerd, was er meer variatie tussen stadia: in de S1 waren de verschillen vrij klein, met 9-Decenol als de meest upregulerende metaboliet (log2-voudige verandering = 2,7; vergeleken met LR). Analyses van de S2 en S3 leverden zeer congruente resultaten, met fenol (respectievelijk 3,7 en 3.2) als de meest upregulated metaboliet. In de S4 was indoxylsulfaat (4.7)de topmetaboliet in LB. Er werd een metabole verschuiving waargenomen in de rijpe vrucht (S5), waar de lijst van metabolieten die in LB werden opgepreguleerd werd aangevuld met stearoylcarnitine (7.1), Methoxyazijnzuur (5.3), s-Methyl-5′-thioadenosine (4.7), lisinopril (4.7), Adenosine 3′, 5 ‘ – cyclisch fosfaat (cAMP) (4.7), enz. Andere metabolieten die in hoge mate in LR worden gereguleerd (behalve Fructose-1-fosfaat) waren naringine (6.2), lauroyl-CoA (4.8), L-Phneylalanine (4.6), enz.