technologia sekwencjonowania Illumina wykorzystuje generowanie klastrów i sekwencjonowanie przez syntezę (SBS) Chemia sekwencjonować miliony lub miliardy klastrów na komórce przepływowej, w zależności od platformy sekwencjonowania. Podczas SBS chemistry, dla każdego klastra, wywołania bazy są wykonywane i przechowywane dla każdego cyklu sekwencjonowania przez oprogramowanie do analizy w czasie rzeczywistym (RTA) na przyrządzie. RTA przechowuje dane wywołania bazowego w postaci indywidualnych plików wywołania bazowego (lub BCL). Po zakończeniu sekwencjonowania, wywołania bazowe w plikach BCL muszą być przekonwertowane na dane sekwencji. Proces ten nazywa się BCL do konwersji FASTQ.
plik FASTQ to plik tekstowy zawierający dane sekwencji z klastrów, które przepuszczają filtr w komórce przepływowej (aby uzyskać więcej informacji na temat filtra przepuszczającego klastry, zobacz sekcję „Informacje dodatkowe” tego biuletynu). Jeśli próbki były multipleksowane, pierwszym krokiem w generowaniu plików FASTQ jest demultipleksowanie. Demultipleksowanie przypisuje klastry do próbki na podstawie sekwencji indeksowej klastra. Po demultipleksowaniu zmontowane sekwencje są zapisywane do plików FASTQ na próbkę. Jeśli próbki nie były multipleksowane, etap demultipleksowania nie występuje, a dla każdego pasa komórek przepływowych wszystkie klastry są przypisane do jednej próbki.
dla pojedynczego odczytu tworzony jest jeden plik FASTQ odczytu 1 (R1) dla każdej próbki na pas komórki przepływowej. Dla sparowanego przebiegu końcowego, dla każdej próbki dla każdego pasa tworzony jest jeden plik R1 i jeden Read 2 (R2) FASTQ. Pliki FASTQ są kompresowane i tworzone z rozszerzeniem *.fastq.gz.
jak wygląda plik FASTQ?
dla każdego klastra, który przechodzi filtr, pojedyncza sekwencja jest zapisywana do odpowiedniego pliku próbki R1 FASTQ, a dla sparowanego końca, pojedyncza sekwencja jest również zapisywana do pliku próbki R2 FASTQ. Każdy wpis w pliku FASTQ składa się z 4 linii:
- identyfikator sekwencji z informacjami o przebiegu sekwencjonowania i klastrze. Dokładna zawartość tej linii różni się w zależności od używanego oprogramowania do konwersji BCL na FASTQ.
- Sekwencja (wywołanie bazy; A, C, T, G i N).
- separator, który jest po prostu znakiem plus ( + ).
- oceny jakości połączeń bazowych. Są to kodowane Phred +33, przy użyciu znaków ASCII do reprezentowania liczbowych wyników jakości.
oto przykład pojedynczego wpisu w pliku R1 FASTQ:
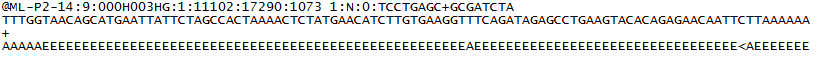
bardziej szczegółowe informacje na temat formatu FASTQ można znaleźć tutaj.
jak wyświetlić plik FASTQ
pliki FASTQ mogą zawierać do milionów wpisów i mogą mieć kilka megabajtów lub gigabajtów, co często czyni je zbyt dużymi, aby je otworzyć w normalnym edytorze tekstu. Ogólnie rzecz biorąc, nie jest konieczne przeglądanie plików FASTQ, ponieważ są to pośrednie pliki wyjściowe używane jako dane wejściowe dla narzędzi, które wykonują analizę niższego szczebla, takich jak wyrównanie do odniesienia lub de novo assembly.
jeśli chcesz wyświetlić plik FASTQ w celu rozwiązywania problemów lub z ciekawości, będziesz potrzebował edytora tekstu, który może obsługiwać bardzo duże pliki, lub dostępu do systemu Unix lub Linux, w którym duże pliki można przeglądać za pomocą wiersza poleceń.
jak wygenerować pliki FASTQ
generowanie plików FASTQ jest pierwszym krokiem dla wszystkich przepływów pracy analizy używanych przez MiSeq Reporter na MiSeq i Local Run Manager na MiniSeq. Po zakończeniu analizy pliki FASTQ znajdują się w <folderze run>\Data\Intensities\BaseCalls na MiSeq i <folderze wyjściowym>\Alignment_#\<podfolderze>\FASTQ na MiniSeq.
dla wszystkich uruchomień przesyłanych do centrum sekwencji BaseSpace generowanie plików FASTQ następuje automatycznie po całkowitym przesłaniu biegu, a pliki FASTQ są używane jako dane wejściowe dla różnych aplikacji do analizy w centrum sekwencji BaseSpace. Na BaseSpace Sequence Hub możesz znaleźć pliki FASTQ w projektach powiązanych z Twoim run.
oprogramowanie do konwersji bcl2fastq może być używane do generowania plików FASTQ z danych generowanych na wszystkich obecnych systemach sekwencjonowania Illumina.
aby uzyskać informacje na temat różnych ustawień, które można zastosować podczas generowania plików FASTQ, zapoznaj się z poniższymi instrukcjami obsługi oprogramowania.
- Miseq Reporter
Local Run Manager
bcl2fastq
dodatkowe informacje
- opis i wymagania dotyczące filtrowania klastrów można znaleźć w sekcji 1.5.8 kursu szkoleniowego MiSeq: Imaging and Base Calling online.
- Zobacz 2-kanałową technologię SBS, aby uzyskać więcej informacji na temat połączeń bazowych w systemach NovaSeq, NextSeq 500/550 i MiniSeq.
- Zobacz technologię sekwencjonowania Illumina, aby uzyskać więcej informacji na temat wywoływania bazy w systemach MiSeq i HiSeq.