vă ajutăm să luați o decizie în cunoștință de cauză
- ¿ce este screening-ul purtător de fibroză chistică?
- ce este fibroza chistică?
- ce este un purtător de fibroză chistică?
- cine ar trebui să fie testat pentru purtătorii de fibroză chistică?
- ¿cum sunt detectați purtătorii de fibroză chizică?
- ce înseamnă o detecție negativă?
- ce înseamnă o detectare pozitivă?
- ce opțiuni de planificare familială sunt disponibile?
- pe scurt…
¿ce este screening-ul purtător de fibroză chistică?
bolile moștenite sau genetice, cum ar fi fibroza chistică (abreviată CF), sunt transmise de la părinte la copil. Acest lucru se întâmplă chiar dacă niciunul dintre părinți nu are boala. Pentru ca un copil să moștenească CF, ambii părinți trebuie să aibă o genă modificată care provoacă CF, adică ambii părinți trebuie să fie purtători ai unei gene modificate pentru CF.
detectarea purtătorului CF vă indică probabilitatea sau riscul de a purta o genă CF modificată. Screening-ul purtător vă poate spune, de asemenea, cât de probabil sunteți să aveți un copil cu CF. Cu toate acestea, screening-ul purtător nu vă poate spune dacă copilul dumneavoastră va avea CF. Sunt necesare dovezi suplimentare în acest scop. Această broșură vă va ajuta să înțelegeți exact ce informații pot fi obținute de la un screening de transport CF și modul în care aceste informații vă pot ajuta să vă planificați familia.
ce este fibroza chistică?
CF este o boală moștenită care apare cel mai adesea la persoanele ale căror strămoși provin din nordul și vestul Europei. În caucazieni din Europa de Nord și descendenți ai evreilor Askenazi, aproximativ 1 copil din 3.000 de nașteri vor prezenta CF. Oamenii din alte grupuri etnice au, de asemenea, CF, dar nu la fel de des. De exemplu, 1 din 8.500 de americani hispanici, 1 din 15.000 de afro-americani și doar 1 din 35.000 de americani asiatici se vor naște cu CF.
persoanele cu CF au niveluri ridicate de sodiu și clorură (sare) în transpirație. Mai important, prezența mucusului gros și lipicios în plămâni provoacă tuse persistentă și respirație șuierătoare, precum și infecții pulmonare frecvente, inclusiv pneumonie. Unii copii au cantități foarte mici de enzime pancreatice și nu pot descompune alimentele în mod normal pentru a extrage nutrienții de care au nevoie pentru a crește. Toate aceste simptome pot fi ușoare sau severe.
infertilitatea (incapacitatea de a avea copii) este, de asemenea, frecventă la persoanele cu CF, în special la bărbați. Deși bărbații cu CF produc spermă, ei se nasc aproape întotdeauna fără vas deferens, tuburile care transportă sperma din testicule. Această afecțiune se numește absența bilaterală congenitală a vaselor deferente (abreviată ABCVD). Deoarece sperma nu poate călători din testicule, acești bărbați nu eliberează sperma în timpul sexului și, prin urmare, sunt sterili. ABCVD poate apărea la bărbații care nu au alte simptome CF, precum și la cei care le au. Pe de altă parte, multe femei cu CF pot avea copii, deși unele au scăzut fertilitatea.
deși CF nu are leac, există unele tratamente care cresc foarte mult speranța de viață a pacienților cu CF. De exemplu, problemele digestive sunt adesea tratate cu diete speciale bogate în proteine, calorii și vitamine. În plus, capsulele sunt luate cu fiecare masă pentru a înlocui enzimele pancreatice lipsă. Simptomele pulmonare sunt tratate cu terapie fizică care uneori poate elimina mucusul din plămâni. Se utilizează, de asemenea, antibiotice și alte medicamente. În ciuda tratamentului, jumătate dintre persoanele născute cu CF mor înainte de vârsta de 37 de ani, în principal din cauza bolilor pulmonare.
ce este un purtător de fibroză chistică?
pentru majoritatea caracteristicilor moștenite, primim 1 genă de la mama noastră și 1 genă de la Tatăl nostru. Persoanele care primesc 1 genă CF modificată și 1 genă normală sunt numite purtători CF deoarece” poartă ” o genă CF modificată, dar nu au CF. Acești purtători au o șansă de 50% de a transmite gena CF modificată fiecăruia dintre copiii lor. Cu toate acestea, pentru ca un copil să moștenească CF, el sau ea trebuie să primească o genă CF modificată de la ambii părinți. Un copil nu poate moșteni CF atunci când doar 1 părinte este purtător. Persoanele cu CF sunt, de asemenea, purtători, deoarece” poartă ” 2 gene CF modificate. Acești oameni vor transmite o genă CF modificată fiecăruia dintre copiii lor.
cine ar trebui să fie testat pentru purtătorii de fibroză chistică?
aceasta este o decizie pe care trebuie să o luați dumneavoastră, partenerul dumneavoastră și medicul dumneavoastră. Detectarea transportatorului este oferită:
- toate cuplurile care așteaptă un copil
- cei care planifică o sarcină
- Cuplurile în care unul dintre membrii cu FC
- persoanele cu antecedente familiale de FC, ca cei care au deja un copil cu FC sau cei care au o rudă directă cu FC
bărbați cu abcvd (vezi secțiunea de mai sus: „Ce este fibroza chistică?”) sunt adesea purtători sau au CF. Deoarece acești bărbați pot avea încă copii printr-o procedură specială în care extrag sperma direct din testicule, ei și partenerii lor ar putea dori să fie supuși screeningului.
poate doriți să luați în considerare riscul dvs. de transport der. Riscul se bazează pe istoricul personal și familial al CF și pe frecvența CF în grupul dvs. etnic. Tabelul 1 enumeră riscul purtător pentru mai multe grupuri. De asemenea, poate doriți să luați în considerare capacitatea testului de a descoperi gene CF modificate care pot apărea în grupul dvs. etnic, așa cum se arată în tabelul 2 și descris în secțiunea următoare („cum se face detectarea purtătorilor de fibroză chistică?”) Amintiți-vă că screening-ul purtător oferă informații despre șansele copilului dvs. de a moșteni CF, dar nu vă poate spune dacă veți transmite sau nu CF copilului dumneavoastră.
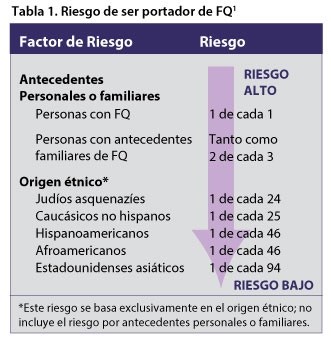
¿cum sunt detectați purtătorii de fibroză chizică?
au fost descoperite peste 1.700 de modificări ale genei CF. Cu toate acestea, testul de screening identifică doar cele mai frecvente modificări. Prin urmare, testul este mai bun pentru găsirea transportatorilor CF în unele grupuri etnice decât în altele. De exemplu, testul poate detecta 88% din modificările găsite în grupul Caucazian non-Hispanic, dar doar 72% din modificările găsite în grupul Hispanic American. Tabelul 2 prezintă specificitatea cu care testul sonor al laboratoarelor Quest poate găsi gene CF modificate în fiecare grup etnic.

detectarea purtătorilor CF se face pe o mică probă de sânge. În timpul testului, Laboratorul va afla dacă purtați una dintre cele mai frecvente modificări ale genei CF. Medicul dumneavoastră ar trebui să trimită informații despre rasa, etnia și orice istoric personal sau familial de CF la laborator pentru a ajuta la interpretarea rezultatelor.
ce înseamnă o detecție negativă?
după cum se arată în tabelul 1, riscul dumneavoastră de FC se bazează pe etnia dumneavoastră și pe istoricul personal sau familial. CF carrier screening oferă informații suplimentare care vă modifică riscul ” înainte de testare.”O detectare negativă înseamnă că laboratorul nu a găsit nicio modificare a genelor CF, astfel încât riscul dvs. real de a fi purtător CF este mai mic decât riscul dvs. pre-test. De asemenea, este mai puțin probabil să aveți un copil cu CF. Deoarece testul de screening găsește doar cele mai frecvente modificări ale genei CF, un screening negativ nu garantează că nu este un purtător.
ce înseamnă o detectare pozitivă?
o detectare pozitivă înseamnă că laboratorul a găsit o schimbare în 1 din cele 2 gene CF și că sunteți un purtător. Există o șansă de 50% să transmiteți gena modificată copilului dumneavoastră. Chiar dacă transmiteți gena modificată copilului dvs., aceasta nu înseamnă neapărat că copilul dvs. va avea CF. Șansele copilului dvs. de a avea CF depind, de asemenea, dacă partenerul dvs. are sau nu CF.
dacă partenerul dvs. are un screening negativ pentru purtătorii CF, șansele dvs. de a avea un copil cu CF sunt mai mici decât dacă screeningul partenerului dvs. a fost pozitiv; cu toate acestea, dacă screeningul partenerului dvs. a fost pozitiv; cu toate acestea, există o șansă de 50%, sau 1 din 2, ca copilul dvs. să fie purtător CF. Singurul mod în care tu și partenerul dvs. ați putea avea un copil cu CF este dacă partenerul dvs. are o schimbare rară în 1 dintre genele CF care nu a fost detectată la test.
șansele ca acest lucru să se întâmple depind de rasa și istoricul familiei partenerului tău. Medicul sau consilierul genetic vă poate oferi informații mai specifice.
dacă partenerul dvs. este și purtător, atunci există o șansă de 25%, sau 1 din 4, ca copilul dvs. să aibă CF. Există o șansă de 50%, sau 1 din 2, ca copilul dvs. să nu aibă CF, ci să fie purtător. În cele din urmă, există o șansă de 25%, sau 1 din 4, ca copilul dvs. să nu fie nici măcar un purtător.
dacă partenerul tău are CF, atunci există o șansă de 50%, sau 1 din 2, ca copilul tău să aibă CF. Există, de asemenea, o șansă de 50% ca copilul dvs. să nu aibă CF.
amintiți-vă că riscul descris mai sus va fi același pentru fiecare copil pe care îl concepeți și partenerul dvs. (adică pentru fiecare sarcină).
de asemenea, amintiți-vă că screening-ul pentru purtătorii de CF vă poate spune doar riscul ca copilul dumneavoastră să aibă CF; nu poți determina dacă vreunul dintre copiii tăi va avea CF.
ce opțiuni de planificare familială sunt disponibile?
dacă screeningul transportatorului este negativ și nu aveți membri apropiați ai familiei cu FC, vă puteți planifica familia știind că este foarte puțin probabil să aveți un copil cu FC. Acestea sunt adevărate chiar dacă partenerul dvs. este purtător de CF, deoarece sunt necesare 2 gene modificate pentru a avea un copil cu CF. Cu toate acestea, amintiți-vă că nu există garanții că copilul dvs. nu va avea CF.
dacă tu și partenerul tău aveți CF, poate doriți să știți cum trăiți cu o persoană cu CF și grija de care au nevoie. De asemenea, poate doriți să știți despre procedurile medicale care vă spun dacă un copil nenăscut a moștenit CF. De exemplu, eșantionarea villusului corionic ia o probă de țesut placentar între săptămânile 10 și 12 de sarcină. Acest eșantion este apoi analizat pentru modificările găsite în genele CF ale părinților. O altă procedură, numită amniocenteză, ia o probă de lichid amniotic, lichidul care înconjoară și protejează copilul, între săptămânile 14 și 18 de sarcină. Acest fluid conține celule care au ieșit de pe pielea bebelușului. Aceste celule sunt testate pentru genele modificate ale părinților CF. De asemenea, poate doriți să știți despre alte metode de concepere, cum ar fi utilizarea unui ovul sau a spermei de la un donator care nu este probabil să poarte CF sau utilizarea diagnosticului genetic pre-implant împreună cu fertilizarea in vitro. În cele din urmă, ați putea dori să exploreze opțiunea de adopție. Medicul sau consilierul genetic vă poate ajuta să aflați mai multe despre aceste opțiuni și despre riscurile și beneficiile asociate acestora. Informațiile vă vor ajuta să luați cele mai bune decizii pentru dvs. și familia dvs.
pe scurt…
Screening-ul pentru purtătorii de fibroză chistică
- determină riscul de a purta o genă modificată care poate provoca FC
- determină riscul de a transmite acea genă copilului dumneavoastră
- determină riscul copilului dumneavoastră de FC dacă dumneavoastră și partenerul dumneavoastră faceți screening
un screening negativ înseamnă că
- niciuna dintre genele FC comune nu a fost găsită în proba de sânge
- este mult mai puțin probabil să transmiteți copilului dumneavoastră o genă cf modificată
- este mult mai puțin probabil să aveți un copil cu CF
amintiți-vă că un screening negativ nu garantează că nu sunteți purtător și nu puteți transmite copilului dumneavoastră o genă CF modificată.
o detectare pozitivă înseamnă că
- este un purtător CF
- ar putea transmite această genă CF modificată copilului dumneavoastră
- copilul dumneavoastră ar putea avea CF dacă partenerul dumneavoastră este, de asemenea, un purtător
detectarea purtătorilor CF nu vă poate spune cu siguranță dacă veți avea sau nu copil cu CF. Cu toate acestea, screening-ul transportatorului vă va oferi informații importante care vă vor ajuta să luați cele mai bune decizii posibile pentru dvs. și familia dvs.
referințe:
1. Richards CS, Bradley LA, Amos J, și colab. Standarde și orientări pentru testarea mutației CFTR. Genet Med. 2002:4:379-391.
2. Colegiul American de Genetică Medicală. Standarde tehnice și orientări pentru testarea mutațiilor CFTR. 2006 ed. http://www.acmg.net/Pages/ACMG_Activities/stds-2002/cf.htm#Table1. Accesat La 10 Mai 2010.
3. Comitetul pentru Genetică, Colegiul American de Obstetrică și Ginecologie. Avizul Comitetului ACOG. Numărul 325, Decembrie 2005. Actualizare privind screeningul purtător pentru fibroza chistică. Obstetret Gynecol. 2005;106:1465-1468.