Illumina sekvenseringsteknik använder klustergenerering och sekvensering genom syntes (SBS) kemi för att sekvensera miljoner eller miljarder kluster på en flödescell, beroende på sekvenseringsplattformen. Under SBS kemi, för varje kluster, bas samtal görs och lagras för varje cykel av sekvensering av realtidsanalys (rta) programvara på instrumentet. RTA lagrar basanropdata i form av enskilda basanrop (eller BCL) filer. När sekvenseringen är klar måste basanropen i BCL-filerna konverteras till sekvensdata. Denna process kallas BCL till FASTQ-konvertering.
en FASTQ-fil är en textfil som innehåller sekvensdata från kluster som passerar filter på en flödescell (för mer information om kluster passerar filter, Se avsnittet ”Ytterligare information” i denna bulletin). Om prover multiplexerades är det första steget i FASTQ-filgenerering demultiplexering. Demultiplexering tilldelar kluster till ett prov, baserat på klustrets indexsekvens(er). Efter demultiplexering skrivs de sammansatta sekvenserna till FASTQ-filer per prov. Om prover inte multiplexerades inträffar inte demultiplexeringssteget, och för varje flödescellsfält tilldelas alla kluster till ett enda prov.
för en enkelläsning skapas en Read 1 (R1) FASTQ-fil för varje prov per flödescellsfält. För en parad slutkörning skapas en R1 och en Read 2 (R2) FASTQ-fil för varje prov för varje körfält. FASTQ-filer komprimeras och skapas med tillägget *.fastq.gz.
Hur ser en FASTQ-fil ut?
för varje kluster som passerar filter skrivs en enda sekvens till motsvarande provets R1 FASTQ-fil, och för en parad slutkörning skrivs också en enda sekvens till provets R2 FASTQ-fil. Varje post i en FASTQ filer består av 4 rader:
- en sekvensidentifierare med information om sekvenseringskörningen och klustret. Det exakta innehållet i denna rad varierar beroende på BCL till FASTQ konvertering programvara som används.
- sekvensen (basen anropar; A, C, T, G och N).
- en separator, som helt enkelt är ett plus (+) tecken.
- basen samtalskvalitet poäng. Dessa är Phred + 33 kodade med ASCII-tecken för att representera de numeriska kvalitetsresultaten.
här är ett exempel på en enda post i en R1 FASTQ-fil:
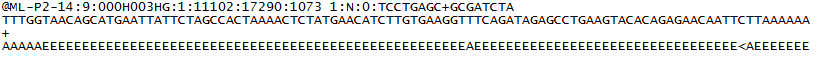
mer detaljerad information om FASTQ-formatet finns här.
hur man visar en FASTQ-fil
FASTQ-filer kan innehålla upp till miljoner poster och kan vara flera megabyte eller gigabyte i storlek, vilket ofta gör dem för stora för att öppnas i en vanlig textredigerare. I allmänhet är det inte nödvändigt att visa FASTQ-filer, eftersom de är mellanliggande utdatafiler som används som ingång för verktyg som utför nedströmsanalys, såsom anpassning till en referens eller de novo-montering.
om du behöver visa en FASTQ-fil för felsökningsändamål eller av nyfikenhet behöver du antingen en textredigerare som kan hantera mycket stora filer eller tillgång till ett Unix-eller Linux-system där stora filer kan ses via kommandoraden.
hur man genererar FASTQ-filer
FASTQ-filgenerering är det första steget för alla analysarbetsflöden som används av MiSeq Reporter på MiSeq och Local Run Manager på MiniSeq. När analysen är klar finns FASTQ-filerna i <kör mapp> \ Data \ Intensities\BaseCalls på utmatningsmappen MiSeq och <>\Alignment_#\<undermapp>\Fastq på MiniSeq.
för alla körningar som laddas upp till BaseSpace Sequence Hub sker FASTQ-filgenerering automatiskt efter att körningen är helt uppladdad och FASTQ-filerna används som inmatning för de olika analysapparna på BaseSpace Sequence Hub. På BaseSpace Sequence Hub kan du hitta dina FASTQ-filer i projektet / projekten som är associerade med din körning.
den bcl2fastq konvertering programvara kan användas för att generera FASTQ filer från data som genereras på alla aktuella Illumina sekvenseringssystem.
för information om de olika inställningarna som kan tillämpas under FASTQ-filgenerering, se användarhandböckerna för programvaran nedan.
- MiSeq Reporter
Local Run Manager
bcl2fastq
ytterligare information
- en beskrivning och krav för kluster att passera filter kan hittas i avsnitt 1.5.8 i MiSeq: Imaging och Base Calling online utbildning.
- se 2-kanals SBS-teknik för mer information om basanrop på NovaSeq, NextSeq 500/550 och MiniSeq-system.
- se Illumina-sekvenseringsteknik för mer information om basanrop på MiSeq-och HiSeq-system.