foram coletados frutos de barbarum L. e L. ruthenicum em cinco estágios de desenvolvimento, desde jovens de frutas (≈10 dias pós-floração), para adultos (ripe), frutas (34-45 dias pós-floração), e estudou seu transcriptoma e metaboloma.
- RNA-seq de novo assembly and functional annotation of unigenes
- Lycium barbarum (LB): pairwise intermediária análises
- LB Transcriptoma
- Metaboloma de LB
- Lycium rutenicum (LR): pairwise intermediária análises
- LR Transcriptoma
- Metaboloma LR
- Intra-análise comparativa dos DEGs em diferentes estágios de desenvolvimento
- > Total DEGs durante o desenvolvimento de frutos
- Transcriptoma – caminhos
- Transcriptoma – genes individuais
- Metabolome – pathways
- Metabolome – metabolitos individuais
RNA-seq de novo assembly and functional annotation of unigenes
we prepared a total of 30 cDNA libraries from fruits of L. barbarum and L. rutenicum, with three biological replicates (three fruits from three trees) at each time point: 2 species × 5 time-points × 3 biological replicates. As amostras foram rotuladas LB/LR (1-5)-(1-3), onde LB é L. barbarum e LR é l. rutenicum, 1-5 são estágios de desenvolvimento de frutos( S1-S5), e 1-3 São amostras individuais (replicados biológicos); assim, por exemplo, LB1-1 representa L. barbarum, 1.a fase de desenvolvimento amostrada (S1), amostra de fruta n. o 1 (de três). Nós geramos mais de 1,72 bilhões de leituras par-end para essas 30 bibliotecas cDNA, correspondendo a uma média de 57,2 milhões de leituras por amostra (Conjunto de dados suplementar S1). A rigorosa avaliação da qualidade e filtragem de dados rendeu um total de 801.766 leituras de alta qualidade com o comprimento médio de 730 e N50 de 1107 bp (Quadro 1). Finalmente, um total de 326.276 unigenes com o comprimento médio de 596 bp e N50 de 847 bp foram obtidos a partir das transcrições (Quadro 1). Os coeficientes de correlação para os dados de ARN-seq para as 30 amostras indicam uma boa consistência dos resultados entre os replicados biológicos (Fig. 2).

Heatmap of correlation coefficients for RNA-seq data for 30 samples of L. barbarum (LB) and L. ruthenicum (LR) fruits at five different developmental stages. As amostras são rotuladas LB / R1–5_1-3, em que LB é L. barbarum, LR é L. ruthenicum, 1-5 são fases de desenvolvimento de frutas, e 1-3 amostras individuais. As amostras foram agrupadas por clustering hierárquico; dendrogramas acima e à esquerda do mapa de calor indicam relação de amostras.
Entre todos os 326,276 unigenes consultado contra bancos de dados públicos, de um total de 193,021 (59.15%) de correspondência de genes e/ou proteínas em pelo menos um banco de dados, e 12,171 (3.73%) foram anotados em todos os bancos de dados. O maior número de unigenes (149,863, 45,93%) foi anotado na base de dados NT, e o menor número (24,017; 7,36%) na base de dados KOG.
Lycium barbarum (LB): pairwise intermediária análises
LB Transcriptoma
O maior número de DEGs foram identificados em todas as comparações em pares da 1ª fase e na 2ª vs. 5ª etapa de comparação (todos DEGs; Tabela 2). Os menores números foram identificados nas comparações do 3º vs. 4º e 4º vs. 5º estágios (255-257). A análise Heatmap do DEGs na LB mostra que conjuntos bastante diferentes de genes foram altamente regulados nos estágios iniciais de Desenvolvimento (1 + 2) e em estágios posteriores (3 a 5) (Fig. 3A). A análise da relação da amostra indica que as amostras podem ser divididas em dois clades (fases 1 + 2 e 3 + 4 + 5), com o último clado subdividido em dois clados: fases 3 + 4 e fase 5. A análise da classificação funcional das Gegg intra-sectorial identificou 15 vias significativamente (P < 0, 05) regulamentadas diferencialmente entre diferentes fases de desenvolvimento (conjunto de dados suplementar S2). Particularmente fortemente regulamentados diferentemente foram “transdução de sinais de hormona vegetal”, ” biossíntese fenilpropanóide (B.)”, ” metabolismo do ácido linoleico (m.)”, ” amido e sacarose M.”, e ” zeatin B. “(Fig. 3A).

os mapas e funcional caminho análises de diferencialmente expressos genes (DEGs) em Lycium barbarum (painel A) e L. ruthenicum (painel B) frutos. Heatmaps foram gerados por uma análise hierárquica de DEGs (eixo y) e amostras individuais (eixo x), onde dendrogramas acima e à esquerda do heatmap indicam relação de amostras. As amostras são rotuladas LB / R_1–5_1–3, em que o acrónimo de espécie (LB ou LR) é seguido pela fase de desenvolvimento dos frutos (1-5) e pelo número da amostra (1-3). As análises da via Kegg intraspecífica do DEGs em todas as cinco fases de desenvolvimento das duas espécies são mostradas à direita dos rotores de calor. Apenas as 15 melhores vias enriquecidas estão listadas. o valor de q é um valor p ajustado pelo FDR.
para uma análise mais aprofundada dos dados, focamos na comparação das vias mais significativamente regulamentadas em fases sucessivas de desenvolvimento. Na primeira comparação emparelhada (S1 vs S2), o “fenilpropanóide B.” Foi a via de regulação mais diferenciada, seguida de “amido e sacarose m.” (Fig. 4). Foram identificados números muito elevados de DEGs (>100) em ambas as vias. Um resultado semelhante foi observado na seguinte comparação emparelhada, S2 vs. S3, mas apesar do número relativamente elevado de DEGs (>80) ‘amido e sacarose M.”exibiu um valor de q um pouco menor. Na comparação S3 vs. S4, “fixação de carbono em fotossintéticos, organismos” foi a via mais significativamente regulada, mas o número de genes foi muito menor. No último par, S4 vs. S5, ‘zeatina b.’, icariin b.’, ácido graxo b.’ e ‘galactose m.’ foram os que mais significativamente regulamentado caminhos, mas nenhum dos percursos exibidos mais de dois DEGs.

LB transcriptoma: pairwise intermediária KEGG via metabólica de enriquecimento. (A) Fase 1 vs. Fase 2. (B) Fase 2 vs. Fase 3. (C) Fase 3 vs. Fase 4. (D) Fase 4 vs. fase 5. Top 15 (ou todos se total <15) vias significativamente enriquecidas são mostradas. A cor da barra representa a magnitude do valor q (um valor p ajustado pelo FDR). O gráfico de cores é mostrado na figura.
Metaboloma de LB
conduzimos comparações emparelhadas de fases de desenvolvimento para identificar o enriquecimento de metabolitos entre todos os pares de fases. O número de metabolitos regulados diferencialmente em comparações emparelhadas das fases de desenvolvimento na LB variou entre 66 (fases 3 vs 4) e 129 (fases 2 vs 5). O maior número de comparações em fases sucessivas foi observado entre a primeira e a segunda fase (117). Estas foram atribuídas a um grande número de vias; com o maior número de metabolitos atribuídos a “B. de metabolitos secundários” (também o valor p Mais baixo), seguido de “digestão e absorção proteicas” e “B. de aminoácidos” (Fig. 5; Números Suplementares). Na comparação sucessiva dos pares de estágios (S2 vs. S3), “transportadores ABC” foi o caminho mais significativamente enriquecido, seguido de “purine M.”. Na comparação de pares S3 vs. S4 ,o “microbiano M. em diversos ambientes” foi a via mais significativamente enriquecida, seguida pelo “carbapenem M.”. Na comparação dos pares S4 vs. S5, um número relativamente grande de vias exibiu resultados semelhantes (dois metabolitos e valores-p semelhantes), mas notável é o aparecimento de “isoflavonóide B.” e “flavonóide B.” entre eles (Fig. 5; Números Suplementares).

dados metabolómicos LB: enriquecimento da via metabólica de KEGG em paralelo. (A) Fase 1 vs. Fase 2. (B) Fase 2 vs. Fase 3. (C) Fase 3 vs. Fase 4. (D) Fase 4 vs. fase 5. Factor rico é a razão entre o número de metabolitos significativamente regulamentados na via e o número total de metabolitos anotados nessa via (intervalo = 0 a 1, 0). O tamanho do ponto representa o número de metabolitos significativamente enriquecidos na Via correspondente, e a cor do ponto representa o valor P (ambas as legendas mostradas na figura).
no que respeita a cada metabólitos, em S1 vs. S2 comparação, a lista de metabólitos upregulated em S1 foi coberta por Trealose, Glactinol e L-Málico (todos ≈20 log2FC). Ácido oleico, ácido 2-Oxoadípico e ácido esteárico foram os metabolitos mais altamente re-regulados em S2 (todos ≈15 a 18 log2FC). A lista dos metabolitos mais altamente desregulados em S2 em comparação com S3 foi completada por dihidroxiacetona, Lisopc(18:1(9Z)), e adenina (todos ≈16 a 17.5 log2FC). A lista dos metabolitos mais altamente desregulados no S3 (em comparação com o S2) foi completada por trealose, Galactinol e ácido L-málico (todos ≈19 a 20 log2FC). No S3 S4 vs. comparação, a lista dos mais conceituados upregulated metabólitos no S3 foi coberto pela L-Málico ácido DL-Arginina e ácido Oleico (todos ≈18 para 19 log2FC), enquanto a lista dos mais conceituados upregulated metabólitos no S4 foi coberta por D-Manose, N-Acetyllactosamine e LysoPC(18:1(9Z)) em LB (todos ≈16 log2FC). No S4 vs. S5 comparison, the list of most highly upregulated metabolites in the S4 was topped by Trehalose, Palmitic acid, N-Acetilactosamine (all ≈17 to 18 log2FC) in LB. A lista dos metabolitos mais altamente desregulados no S5 foi completada por L-Nroleucina, ácido Antranílico (vitamina L1) e DL-arginina (todos ≈16 a 18 log2FC) (conjunto de dados suplementar S3).
Lycium rutenicum (LR): pairwise intermediária análises
LR Transcriptoma
Heatmap análise dos DEGs em LR mostra que quase que completamente diferentes conjuntos de genes foram altamente upregulated nos primeiros estágios de desenvolvimento (1 + 2) e frutos maduros (fase 5), com uma aparente transcriptomic repor a ocorrer após o segundo estágio (Fig. 3B). A análise da relação da amostra indica que as amostras podem ser divididas em dois clades (fases 1 + 2 e 3 + 4 + 5), com o último clado subdividido em dois clados: fases 3 + 4 e fase 5. A análise da classificação funcional das Gegg intra-sectorial identificou 35 vias significativamente (P < 0, 05) regulamentadas diferencialmente entre diferentes fases de desenvolvimento (conjunto de dados suplementar S2). Particularmente fortemente regulamentados diferentemente foram “fotossíntese”, “ácido linoleico m.”, “alcalóide B isoquinolina B.”, “flavonóide B.” e “amido e sacarose m” (Fig. 3B). Os números mais elevados de DEGs foram identificados na maioria das comparações em pares, a fase 1 (1 vs. 2 foi uma exceção), e na fase 2 vs. 5 de comparação (todos >5,000 DEGs; Tabela 2). De longe, o menor número foi identificado na comparação S3 vs. S4 (39). Na comparação entre S1 e S2 emparelhada ,o “aminoaçúcar e o nucleótido açúcar m.” Foi a via de regulação mais diferenciada, seguida de “ácido linoleico m.” (Fig. 6). Na comparação emparelhada S2 vs. S3, as vias regulamentadas mais significativamente foram o alcalóide B. isoquinolina, a tirosina m., O fenilpropanóide B. e o flavonóide B. Em ambas as comparações emparelhadas, o número mais elevado de DEGs (>15 e >35, respectivamente) foi identificado no “amido e sacarose m.”. No S3 vs. S4 comparison, ‘(alpha -) linoleic acid m. ‘ was the most significantly regulated pathway, but the numbers of genes were much lower. Na comparação S4 vs. S5, o “fenilpropanóide b” (também o maior número de DEGs) e o “ácido linoleico m.” foram as vias regulamentadas mais significativamente.
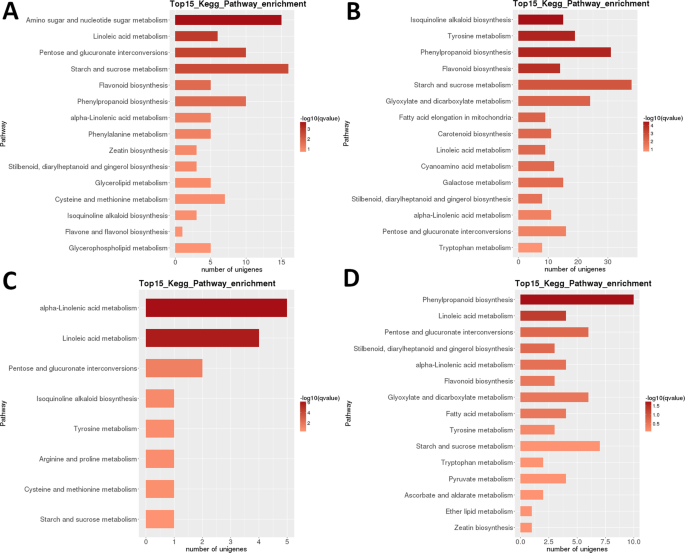
LR transcriptoma: pairwise intermediária KEGG via metabólica de enriquecimento. (A) Fase 1 vs. Fase 2. (B) Fase 2 vs. Fase 3. (C) Fase 3 vs. Fase 4. (D) Fase 4 vs. fase 5. Top 15 (ou todos se total <15) vias significativamente enriquecidas são mostradas. A cor da barra representa a magnitude do valor q (um valor p ajustado pelo FDR, gráfico de cores apresentado na figura).
Metaboloma LR
o número de metabolitos regulamentados diferencialmente em comparações emparelhadas dos estádios de desenvolvimento na LR variou entre 66 (fases 3 vs 4) e 133 (fases 2 vs 5) (Conjunto de dados suplementar S3). Em comparações sucessivas de fases, o maior número foi observado entre a primeira e a segunda fase (117). Estes foram atribuídos a um grande número de vias; com o maior número de metabolitos atribuídos a “b de metabolitos secundários”, seguido de “digestão e absorção proteicas”, “B. de aminoácidos” e “flavonóide b”. Na comparação S2 vs. S3, “transportadores ABC” foi a via mais significativamente enriquecida, seguida de “purine m”. Na comparação de pares S3 vs. S4 ,o “microbiano M. em diversos ambientes” foi a via mais significativamente enriquecida, seguida pelo “carbapenem M.”. No S4 vs. Comparação de pares S5, um número relativamente grande de vias exibiu resultados semelhantes (2 metabolitos e valores-p semelhantes), mas notável é o aparecimento de “isoflavonóide B.” e “flavonóide B.” entre eles (Fig. 7; Números Suplementares).

LR metabolomic de dados: pairwise intermediária KEGG via metabólica de enriquecimento. (A) Fase 1 vs. Fase 2. (B) Fase 2 vs. Fase 3. (C) Fase 3 vs. Fase 4. (D) Fase 4 vs. fase 5. Factor rico é a razão entre o número de metabolitos significativamente regulamentados na via e o número total de metabolitos anotados nessa via (intervalo = 0 a 1, 0). O tamanho do ponto representa o número de metabolitos significativamente enriquecidos na Via correspondente, e a cor do ponto representa o valor P (ambas as legendas mostradas na figura).
no que se refere aos metabolitos individuais (conjunto de dados suplementares S3), no S1 vs. S2 comparação, a lista de metabólitos upregulated em S1 foi coberta por Trealose, Galactinol e L-Málico (≈19 a 21 de log2FC), enquanto que o ácido Oleico, 2-Oxoadipic ácido e ácido Esteárico foram os mais altamente upregulated metabólitos em S2 (≈15-18 log2FC). Na comparação S2 vs. S3, dihidroxiacetona, Indoxilsulfato e N-Acetilactosamina foram os metabolitos mais altamente desregulados no s2 (≈17-19.5 log2FC), e trealose, Galactinol e ácido L-málico (≈19-21 log2FC) no S3. No S3 vs. S4 comparison, l-Malic acid, DL-Arginine and Oleic acid were upregulated in the S3 (≈16-19 log2FC), and 1,7-Dimetilxanthine, D-manose and N-Acetillactosamine (≈15-17 log2FC) in the S4. No S4 vs. S5 comparação, Flavin mononucleotide, Trealose e Isoferulic ácido foram upregulated no S4 (≈18-20 log2FC), considerando que a PG(16:0/18:1(9Z)), D-Prolina, e DL-Arginina foram mais altamente upregulated (todos ≈16 a 18 de log2FC) metabólitos.
Intra-análise comparativa dos DEGs em diferentes estágios de desenvolvimento
> Total DEGs durante o desenvolvimento de frutos
Interespecíficos entre pares fase de comparação (LR1 vs. LB1, LR2 vs. LB2, etc.) mostra que 928 DEGs foram compartilhados por todos os cinco pares (Fig. 8A). O número mais elevado de DEGs foi identificado na fase 3 (3989), e o mais baixo na fase 4 (2825) (Fig. 8B); considerando que o maior número de DEGs únicos a um par foram observados nos estágios 3 (574), 1 e 5 (ambos 554), e o menor na Fase 4 (126) (Fig. 8A). Os números de cima e para baixo-regulado DEGs foram relativamente semelhantes em cada um dos pares fase comparações; por exemplo, no estágio 5, 1668 DEGs foram upregulated e 1670 DEGs foram desativado em LR em comparação com LB (Fig. 8B). No entanto, nas outras quatro fases, o número de genes upregulados foi ligeiramente superior (93 a 189 DEGs).

Genes diferencialmente expressos (DEGs) entre frutos de barbarum L. (LB) e L. ruthenicum (LR). (A) em trechos detalhados (1 a 5) comparações (LB vs. LR). B) O número de DEGs upregulados (vermelhos) e downregulados (verdes) na RL em comparação com o LB em cinco fases de desenvolvimento estudadas.
Transcriptoma – caminhos
Heatmap análise dos DEGs indica que os frutos de duas espécies apresentam muito diferentes perfis de expressão gênica durante todas as fases de desenvolvimento, mas biológicas replica exibiu muito perfis semelhantes, indicando uma quantidade limitada de variabilidade entre os indivíduos em cada estágio de desenvolvimento (Complementar Figuras: Fig. S9). A análise comparativa do enriquecimento da via KEGG mostra que apenas algumas vias foram sistematicamente altamente enriquecidas (em termos de regulação genética) na LR, em comparação com a LB ao longo de todas as cinco fases de desenvolvimento (Fig. 9). Notavelmente, a transdução de sinal de hormônio vegetal (a segunda maior em S1, a oitava mais alta em S2, a mais alta em S3, S4 e S5) e a interação planta-patógeno (a mais alta em S1, a terceira mais alta em S2, a segunda mais alta em s3, A quarta mais alta em S4, e a 15ª mais alta em S5) foram relativamente elevadas em todos os estágios. Phenylpropanoid biossíntese (não no top 15 em S1, o mais alto em S2, 3ª-mais alto no S3, 7, a mais alta S4, 11 a mais alta S5), ubiquinona e outros terpenóides-quinona biossíntese (não no top 15 S1, 6, a mais alta S2, 7, a mais alta S3, 2ª-mais alto em S4, 6, a mais alta S5) também foram relativamente alta, como potenciadas em todas as fases, exceto o primeiro. A via de biossíntese dos flavonóides não foi altamente enriquecida nos estágios iniciais (não no top 15 em S1, 14º mais alto em S2), e altamente enriquecida nos estágios finais (3º A 4º mais alto durante os estágios 3 a 5). o metabolismo do ácido linoleico (alfa -) foi altamente enriquecido em estágios médios (9 ° mais alto em S1, 2 ° mais alto em S2, 5 ° e 6 ° mais alto em s3, 8 ° mais alto em S4, não no top 15 em S5).

análise comparativa do enriquecimento das vias metabólicas de KEGG. Os 15 principais caminhos enriquecidos em L. ruthenicum em comparação com L. barbarum são mostrados à esquerda (Vermelho), e os enriquecidos em L. barbarum em comparação com L. ruthenicum à direita (verde). As fases de desenvolvimento (1-5) são indicadas na figura. o valor de q é um valor p ajustado pelo FDR.
entre os caminhos descregulados em L. ruthenicum em comparação com L. barbarum (Fig. 9) alterações significativas foram observadas entre os estágios iniciais (1 e 2), quando cyanoamino ácido do metabolismo e carotenóide biossíntese foram mais altamente desativado, e em fases tardias (4 e 5), quando o LAÇO medicamentosas em vesicular de transporte, nicotinato e nicotinamida o metabolismo, e a porfirina e a clorofila metabolismo foram consistentemente relativamente alta, desativado.
Transcriptoma – genes individuais
Entre os mais altamente diferencialmente expressos genes, alguns foram estágio de desenvolvimento específico (i.e. altamente diferencialmente regulados somente no início ou no final de estágios de desenvolvimento), mas alguns foram consistentemente altamente diferencialmente regulados ao longo de todas as cinco fases estudadas (Complementar Dataset S4). Vários genes relacionados com a imunidade muito altamente regulados em LR em comparação com LB nos estágios iniciais de desenvolvimento estão entre os exemplos do padrão de expressão específica de estágio de desenvolvimento: chitinase foi o segundo maior DESG em S1 (13,43 vezes), o mais alto em S2 (13,89 vezes), mas em fases posteriores não foi um DESG. Da mesma forma, o receptor EIX 1/2 também foi altamente regulado em estágios iniciais, o mais alto em S1 (13.70) e o segundo mais alto em S2 (10.90), mas também não foi identificado como DEG em estágios posteriores. Alguns genes relacionados ao crescimento também exibiram um padrão de expressão semelhante: a fosfoglicerato cinase (PGK) estava entre os poucos genes mais altamente regulados nos três primeiros estágios (13.14, 12.87 e 12.77, respectivamente), mas também não foi identificado como DEG em fases posteriores. CCR4-a subunidade do complexo de transcrição 7/8 (CNOT7 / 8) também exibiu um padrão de expressão muito semelhante: altamente re-regulada nas três primeiras fases, e não um DESG nas fases 4 e 5. Vários genes associados à biossíntese de flavonóides e fenilpropanóides exibiram um padrão de expressão específica de estágio de desenvolvimento reverso, com expressão relativamente baixa nos estágios iniciais, e muito alta nos estágios posteriores. Exemplos são: di-hidroflavonol bifuncional 4-redutase / flavanona 4-redutase( DFR), que foi ligeiramente regulado em LR em S1 (2.25), não um DEG em S2, altamente upregulado em S3 (7.79), e o terceiro gene mais elevado upregulado em S4 (14.25) e S5 (16.03). Um parálogo deste gene exibia um padrão quase idêntico: ligeiramente ascendente em S1 (2.44), não um DEG em S2, altamente ascendente em S3 (7.40), 6.º DEG mais elevado em S4 (13.26) e 5. º em s5 (14.59). Da mesma forma, icariin 3′,5′-hydroxylase (F3’5 H) não foi uma GRAUS nas duas primeiras fases, altamente upregulated em S3 (6.69), 5º-maior upregulated gene em S4 (13.42), e 4, a mais alta S5 (15.05). O flavonóide o-metiltransferase (OMT) não era um DEG no S1, mas no S2 já exibia um nível de upregulação médio-alto (4.32), pelo S3 já era o terceiro mais elevado DEGREGULADO DEG (13.30), e era o gene mais elevado em S4 (18.73) e S5 (18.10). Leucoantocianidina dioxigenase (LDOX; biossíntese da antocianina) não foi um DEG em S1 e S2, seguido por alta-regulação em fases posteriores (5,63, 9,44, 11,56, respectivamente). Dois parálogos de calcona sintase (CHS e CHS2; biossíntese flavonóide) também não foram altamente regulamentados em S1 e S2 (CHS2: não um DEG, CHS: -1.14 em S1, não um DEG em S2), mas em S3–S5 ambos os genes exibiram um upregulation médio-alto a alto (CHS2: 5.32, 7.84, 6.00; e CHS: 4.67, 7.01, 6.82; respectivamente). Selecionamos esses genes para a análise qPCR, e os resultados são altamente congruentes com os dados RNA-seq (resultados suplementares; conjunto de dados suplementar S5). Finalmente, a citocinina desidrogenase, um gene relacionado com a biossíntese da zeatina, também foi cada vez mais elevada durante os últimos três estágios (2.6–5.7).
no entanto, alguns genes foram consistentemente expressos diferentemente ao longo de todas as cinco fases estudadas. Exemplos também incluiu algumas imunidade-genes relacionados, tais como dois paralogues de glutationa-S-transferase, altamente upregulated em LR em comparação à LIBRA em todas as fases: em 9,38 e 8.58 (todos os valores apresentados como dobra-alterações na respectiva ordem) em S1, 6.30 e 6.34 em S2, o 2º e o 7º maior upregulated DEGs em S3 (14.08 e 12,70), de 2ª e 4ª maior em S4 (15.71 e 14.16) e 2º e 6º maior em S5 (16.48 e 14.40). A proteína RPM1 de resistência às doenças das plantas também foi altamente regulamentada em todos os cinco estágios (S1 = 13.15; S2 = 12.08; S3 = 13.11, S4 = 12.81; S5 = 13:94). Entre os genes consistentemente expressos diferencialmente ao longo de todas as fases de desenvolvimento também estavam alguns relacionados com o metabolismo dos aminoácidos, mas seu padrão foi revertido em comparação com exemplos anteriores: eles exibiram alta regulamentação em LR em comparação com LB. Exemplos são acetil-CoA aciltransferase 1 (AAT1; Valina, Leucina e degradação da isoleucina), com um perfil temporal de cada vez mais baixa regulação, começando de -7,0 no S1 a <−10 vezes nas últimas três fases. Prolina iminopeptidase, associado com arginina e prolina metabolismo, foi altamente desativado em LR em todas as fases: S1 = -9.75, S2 = -10.89 (3ª maior), S3 = -11.05 (de 4 a mais alta), S4 = -10.01, e S5 = -11.98 (3ª-mais alto). Finalmente, 5-methyltetrahydropteroyltriglutamate–homocisteína metiltransferase (metE) foi consistentemente extremamente altamente desativado em LR em todas as fases: 2ª-mais alto em S1 (-11.76), a mais alta em S2 (-11.74), 3ª-mais alto em S3 (-11.43), a mais alta em S4 (-12.36), e 2º, a mais alta S5 (-12.83). Dois replicação do DNA e transcrição associadas a genes também foram altamente desativado em LR em todas as fases: GTP-binding de proteínas nucleares Ran (RAN; -10.0 para -12.0) e replicação fator A1 (RFA1) (-8.0 para -12.0). Alguns crescimento e relacionadas com o stress genes também foram consistentemente altamente desativado em LR: heterogêneo nuclear ribonucleoprotein A1/A3 (hnRNP; -7 a -11) e de choque térmico 70 kDa proteína 1/8 (HSPA1_8) S1 = -4.95, S2 = -8.88, S3 = -11.48 (2ª-mais alto), S4 = -9.36, S5 = -12.89 (o mais alto). Curiosamente, um phenylpropanoid biossíntese relacionadas com o gene, chiquimato hydroxycinnamoyltransferase (HCT), também foi consistentemente altamente desativado em LR: S1 = -6.82, S2 = -8.14, S3 = -11.71 (o mais alto), S4 = -11.00 (3ª maior), S5 = -11.91 (de 4 a mais alta). No entanto, um regulador chave da biossíntese da antocianina, o Fator de transcrição MYB114, foi altamente regulado em LR durante todas as cinco fases de desenvolvimento: 6.11, 4.69, 7.47, 9.05, e 8.95 (S1–S5, respectivamente).
Metabolome – pathways
we also conducted a comparative interespecific stage-wise analysis of metabolic pathways(Fig. 10). Na primeira fase de desenvolvimento (S1), identificamos 39 metabolitos regulados diferencialmente. Entre as 20 principais vias com as quais estes metabolitos foram associados, vários deles foram associados a aminoácidos, mas o número total de metabolitos por via foi relativamente pequeno (1-2), e os valores-P não sugerem um elevado nível de significância (Fig. 10-painel 1). Notavelmente, o metabolismo da vitamina B6 (m) e o m microbiano em diversos ambientes apresentaram valores p comparativamente elevados, factor de enriquecimento (EF) de 1, 0 e 2 metabolitos identificados. No S2, identificamos 58 diferencialmente regulados metabólitos, associado a apenas quatro caminhos: o triptofano m, phenylpropanoid biossíntese (b), b de phenylpropanoids (esses são dois caminhos diferentes no banco de dados KEGG), e a fenilalanina, tirosina e triptofano b (todos os EF = 1.0, 2 e 3 de metabólitos, e p < 0.5; Fig. 10-painel 2). No S3, identificámos 59 metabolitos regulamentados diferencialmente, associados a 19 vias, a maioria com a EF 1, 0, mas relativamente não significativos com valores P (> 0, 5; Fig. 10-painel 3). As vias com um número relativamente elevado de metabolitos (n = 5) foram: digestão e absorção proteicas, b de metabolitos secundários vegetais, b de antibióticos e b de aminoácidos. No S4, identificámos 58 metabolitos regulamentados diferencialmente, associados a um grande número de vias, principalmente com a EF 1, 0, e valores de significância comparativamente elevados (na sua maioria P > 0, 5; Fig. 10-painel 4). Com vias de um número relativamente elevado de metabólitos (n ≥ 3) foram: phenylpropanoid b, fenilalanina, tirosina e triptofano b, glucosinate b, b de alcalóides derivados do chiquimato caminho, e 2-oxocarboxylic m. Nos frutos maduros (S5), identificamos 39 diferencialmente regulados metabólitos, associado a um grande número de vias, mas, principalmente, com baixos valores de ” P ” e apenas 1 metabólito por percurso (Fig. 10-painel 5). As vias com mais de 1 metabolito foram: proteína de digestão e absorção, phenylpropanoid b, absorção de minerais, central de carbono m em câncer, b de metabólitos secundários, b de phenylpropanoids, e aminoacy tRNA b. A análise de componentes principais (PCA) de todos os dados (2 espécies × 5 fases × 5 biológica replica) revelou alta similaridade entre biológica replica (clustering), e corroborado notável variabilidade entre diferentes fruta estágios para ambas as espécies (Fig. 10-painel 6).

análise Comparativa de KEGG via metabólica de enriquecimento. Os 15 principais caminhos enriquecidos em L. ruthenicum em comparação com L. barbarum são mostrados à esquerda (Vermelho), e os enriquecidos em L. barbarum em comparação com L. ruthenicum à direita (verde). As fases de desenvolvimento (1-5) são indicadas na figura. o valor de q é um valor p ajustado pelo FDR.
Metabolome – metabolitos individuais
a lista (Conjunto de dados suplementares S6) dos metabolitos mais altamente regulados diferencialmente entre as duas espécies, exibiu alguma variação entre as cinco fases de desenvolvimento de frutos. Curiosamente, a Frutose 1-fosfato foi o mais altamente upregulated metabólito em LR, face à LIBRA, durante todos os cinco estágios: log2 Fold change = 6.3, 7.6, 7.7, 8.1 e 6.5 (etapas de 1 a 5, respectivamente). No que se refere aos metabolitos altamente regulamentados na LB, verificou-se uma maior variação entre as fases: no S1, as diferenças foram bastante pequenas, com o 9-Decenol como o metabolito mais altamente desregulado (Alteração log2 vezes = 2, 7; em comparação com a LR). As análises do S2 e do S3 produziram resultados altamente congruentes, com o fenol (3.7 e 3.2, respectivamente) como o metabolito mais altamente desregulado. No S4, o sulfato de indoxilo (4, 7) foi o principal metabolito sub-regulado na LB. Uma mudança metabólica foi observado nos frutos maduros (S5), onde a lista de metabólitos upregulated em LB estava coberto por stearoylcarnitine (7.1), Methoxyacetic ácido (5.3), S-Metil-5′-thioadenosine (4.7), lisinopril (4.7), adenosina 3′,5′-fosfato Cíclico (cAMP) (4.7), etc. Outros metabolitos altamente regulamentados na LR (para além da frutose 1-fosfato) foram naringina (6, 2), lauroil-CoA (4, 8), L-Falalanina (4, 6), etc.