Illumina sekvenování technologie využívá clusteru generace a sekvenování syntézou (SBS) chemie sekvence miliony nebo miliardy klastrů na toku buněk, v závislosti na sekvenční platformu. Během SBS chemie, pro každý cluster, základní volání jsou vyrobeny a uloženy pro každý cyklus sekvenování pomocí Real-Time Analysis (RTA) software na přístroji. RTA ukládá data základního volání ve formě jednotlivých souborů základního volání (nebo BCL). Po dokončení sekvenování musí být základní volání v souborech BCL převedena na sekvenční data. Tento proces se nazývá konverze BCL na FASTQ.
FASTQ soubor je textový soubor, který obsahuje sekvence dat z uskupení, které projdou filtrem o průtoku mobilní (pro více informací o uskupení kolem filtru, viz „další informace“ části tohoto bulletinu). Pokud byly vzorky multiplexovány, prvním krokem při generování souborů FASTQ je demultiplexování. Demultiplexing přiřazuje shluky ke vzorku na základě indexové sekvence (y) clusteru. Po demultiplexování jsou sestavené sekvence zapsány do souborů FASTQ na vzorek. Pokud nebyly vzorky multiplexní, demultiplexování krok nenastane, a pro každý tok mobilní lane, všechny clustery jsou přiřazeny do jednoho vzorku.
pro běh s jedním čtením je vytvořen jeden soubor Fastq pro čtení 1 (R1) pro každý vzorek na pruh průtokové buňky. Pro párový konec běhu, jeden R1 a jeden Read 2 (R2) FASTQ soubor je vytvořen pro každý vzorek pro každý pruh. FASTQ soubory jsou komprimovány a vytvořeny s příponou *.fastq.gz.
jak vypadá soubor FASTQ?
pro každý cluster, který prochází filtrem, je jedna sekvence zapsána do souboru R1 FASTQ odpovídajícího vzorku a pro párový běh je také zapsána jedna sekvence do souboru R2 FASTQ vzorku. Každá položka v souborech FASTQ se skládá ze 4 řádků:
- identifikátor sekvence s informacemi o sekvenčním běhu a clusteru. Přesný obsah tohoto řádku se liší v závislosti na použitém softwaru pro konverzi BCL na FASTQ.
- sekvence (základní volání; A, C, T, G A N).
- oddělovač, což je jednoduše znaménko plus ( + ).
- skóre kvality základního hovoru. Jedná se o kód Phred +33, který používá znaky ASCII k reprezentaci číselných skóre kvality.
Zde je příklad jedné položky v R1 FASTQ:
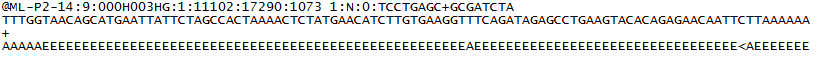
podrobnější informace o FASTQ formátu lze nalézt zde.
jak zobrazit soubor FASTQ
soubory FASTQ mohou obsahovat až miliony záznamů a mohou mít velikost několika megabajtů nebo gigabajtů, což je často činí příliš velkými pro otevření v normálním textovém editoru. Obecně platí, že není nutné zobrazit FASTQ soubory, protože jsou mezilehlé výstupní soubory používané jako vstup pro nástroje, které provádějí následné analýzy, jako je zarovnání k odkazu nebo de novo sestavy.
Pokud potřebujete zobrazit FASTQ souboru pro účely odstraňování potíží, nebo ze zvědavosti, budete potřebovat buď textový editor, který dokáže zpracovat velmi velké soubory, nebo přístup k systému Unix nebo Linux systému, kde velké soubory lze zobrazit pomocí příkazového řádku.
Jak generovat FASTQ soubory
FASTQ generace je prvním krokem pro všechny analýzy pracovní postupy používané MiSeq Reportér na MiSeq a Místní Spustit Správce na MiniSeq. Při dokončení analýzy, FASTQ soubory jsou umístěny v <spustit složku>\Data\Intenzity\BaseCalls na MiSeq a <výstupní složku>\Alignment_#\<podsložka>\Fastq na MiniSeq.
Pro všechny se spouští nahrané na BaseSpace Sekvence Hub, FASTQ generace dochází automaticky po spuštění je zcela nahrán, a FASTQ soubory jsou používány jako vstupní údaje pro různé analýzy aplikace na BaseSpace Sekvence Hub. Na BaseSpace Sequence Hub, můžete najít FASTQ soubory v projektu (y) spojené s vaší běhu.
konverzní software bcl2fastq lze použít ke generování FASTQ soubory z dat generovaných na všech současných sekvenčních systémů Illumina.
informace o různých nastaveních, která lze použít během generování souborů FASTQ, naleznete v uživatelských příručkách softwaru níže.
- MiSeq Reportér
Místní Spustit Správce
bcl2fastq
Další informace
- popis a požadavky pro klastry-pass filtr lze nalézt v oddíle 1.5.8 z MiSeq: Zobrazování a Základní Volání on-line školení.
- viz 2-kanálová technologie SBS pro více informací o volání základny na systémech NovaSeq, NextSeq 500/550 a MiniSeq.
- viz technologie sekvenování Illumina pro více informací o volání základny na systémech MiSeq a HiSeq.