La tecnologia di sequenziamento Illumina utilizza la generazione di cluster e il sequenziamento per sintesi (SBS) per sequenziare milioni o miliardi di cluster su una cella di flusso, a seconda della piattaforma di sequenziamento. Durante la chimica SBS, per ogni cluster, le chiamate di base vengono effettuate e memorizzate per ogni ciclo di sequenziamento dal software di analisi in tempo reale (RTA) sullo strumento. RTA memorizza i dati delle chiamate di base sotto forma di singoli file di chiamata di base (o BCL). Al termine del sequenziamento, le chiamate di base nei file BCL devono essere convertite in dati di sequenza. Questo processo è chiamato conversione da BCL a FASTQ.
Un file FASTQ è un file di testo che contiene i dati di sequenza dei cluster che passano il filtro su una cella di flusso (per ulteriori informazioni sui cluster che passano il filtro, vedere la sezione “informazioni aggiuntive” di questo bollettino). Se i campioni sono stati multiplexati, il primo passo nella generazione di file FASTQ è demultiplexing. Demultiplexing assegna i cluster a un campione, in base alle sequenze di indice del cluster. Dopo il demultiplexing, le sequenze assemblate vengono scritte in file FASTQ per campione. Se i campioni non sono stati multiplexati, la fase di demultiplexing non si verifica e, per ogni corsia di cella di flusso, tutti i cluster vengono assegnati a un singolo campione.
Per una singola esecuzione di lettura, viene creato un file FASTQ di lettura 1 (R1) per ogni campione per corsia di cella di flusso. Per un’esecuzione accoppiata, viene creato un file FASTQ R1 e uno Read 2 (R2) per ogni campione per ogni corsia. I file FASTQ vengono compressi e creati con l’estensione *.veloce.zg.
Che aspetto ha un file FASTQ?
Per ogni cluster che passa il filtro, una singola sequenza viene scritta nel file R1 FASTQ del campione corrispondente e, per un’esecuzione accoppiata, una singola sequenza viene scritta anche nel file R2 FASTQ del campione. Ogni voce in un file FASTQ è composta da 4 righe:
- Un identificatore di sequenza con informazioni sull’esecuzione di sequenziamento e sul cluster. Il contenuto esatto di questa linea varia in base al BCL al software di conversione FASTQ utilizzato.
- La sequenza (le chiamate di base; A, C, T, G e N).
- Un separatore, che è semplicemente un segno più ( + ).
- I punteggi di qualità delle chiamate di base. Questi sono Phred + 33 codificati, utilizzando caratteri ASCII per rappresentare i punteggi di qualità numerica.
Ecco un esempio di una singola voce in un file R1 FASTQ:
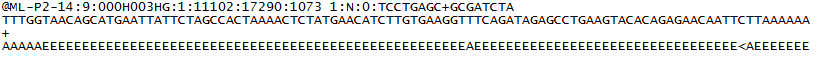
Informazioni più dettagliate sul formato FASTQ possono essere trovate qui.
Come visualizzare un file FASTQ
I file FASTQ possono contenere fino a milioni di voci e possono avere dimensioni di diversi megabyte o gigabyte, il che spesso li rende troppo grandi per essere aperti in un normale editor di testo. Generalmente, non è necessario visualizzare i file FASTQ, perché sono file di output intermedi utilizzati come input per strumenti che eseguono analisi a valle, come l’allineamento a un riferimento o un assembly de novo.
Se è necessario visualizzare un file FASTQ per scopi di risoluzione dei problemi o per curiosità, è necessario un editor di testo in grado di gestire file molto grandi o l’accesso a un sistema Unix o Linux in cui i file di grandi dimensioni possono essere visualizzati tramite la riga di comando.
Come generare file FASTQ
La generazione di file FASTQ è il primo passo per tutti i flussi di lavoro di analisi utilizzati da MISEQ Reporter su MiSeq e Local Run Manager su MiniSeq. Al termine dell’analisi, i file FASTQ si trovano in <esegui cartella>\Data\Intensities\BaseCalls sulla MiSeq e <cartella di output>\Alignment_#\<sottocartella>\Fastq sulla MiniSeq.
Per tutte le esecuzioni caricate su BaseSpace Sequence Hub, la generazione di file FASTQ avviene automaticamente dopo che l’esecuzione è stata completamente caricata e i file FASTQ vengono utilizzati come input per le varie app di analisi su BaseSpace Sequence Hub. Su BaseSpace Sequence Hub, puoi trovare i tuoi file FASTQ nei progetti associati alla tua esecuzione.
Il software di conversione bcl2fastq può essere utilizzato per generare file FASTQ dai dati generati su tutti gli attuali sistemi di sequenziamento Illumina.
Per informazioni sulle diverse impostazioni che possono essere applicate durante la generazione di file FASTQ, consultare le guide utente del software riportate di seguito.
- MISEQ Reporter
Local Run Manager
bcl2fastq
Ulteriori informazioni
- Una descrizione e i requisiti per i cluster per passare il filtro sono disponibili nella sezione 1.5.8 del corso di formazione online MiSeq: Imaging and Base Calling.
- Vedere la tecnologia SBS a 2 canali per ulteriori informazioni sulle chiamate di base sui sistemi NovaSeq, NextSeq 500/550 e MiniSeq.
- Vedere Illumina Sequencing Technology per ulteriori informazioni sulle chiamate di base sui sistemi MiSeq e HiSeq.