Illumina sequencing technology maakt gebruik van clustergeneratie en sequencing door synthese (SBS) chemie om miljoenen of miljarden clusters op een stroomcel te sequencen, afhankelijk van het sequencing platform. Tijdens SBS-chemie worden voor elke cluster basisoproepen gemaakt en opgeslagen voor elke sequencingcyclus door de Real-Time Analysis (RTA) – software op het instrument. RTA slaat de base call data op in de vorm van individuele base call (of BCL) bestanden. Wanneer het rangschikken is voltooid, moeten de basisaanroepen in de BCL-bestanden worden omgezet in sequentiegegevens. Dit proces heet BCL naar fastq conversie.
een fastq-bestand is een tekstbestand dat de sequentiegegevens bevat van de clusters die een filter doorgeven op een stroomcel (voor meer informatie over clusters die een filter doorgeven, zie de sectie “Aanvullende informatie” van dit bulletin). Als de steekproeven werden gemultiplexed, is de eerste stap in fastq-dossiergeneratie demultiplexing. Demultiplexing wijst clusters toe aan een steekproef, die op de opeenvolging(s) van de clusterindex wordt gebaseerd. Na demultiplexing, worden de geassembleerde opeenvolgingen geschreven aan fastq dossiers per steekproef. Als de steekproeven niet werden gemultiplexed, komt de demultiplexingstap niet voor, en, voor elke stromingscelstrook, worden alle clusters aan één enkele steekproef toegewezen.
voor een enkele READ run wordt één Read 1 (R1) FASTQ-bestand aangemaakt voor elk monster per stromingscelstrook. Voor een gepaarde einde run, een R1 en een Read 2 (R2) FASTQ-bestand wordt gemaakt voor elk monster voor elke rijstrook. FASTQ-bestanden worden gecomprimeerd en gemaakt met de extensie *.fastq.gz.
hoe ziet een fastq-bestand eruit?
voor elk cluster dat het filter passeert, wordt een enkele reeks geschreven naar het R1 FASTQ-bestand van het corresponderende monster en, voor een gepaarde run, wordt ook een enkele reeks geschreven naar het R2 FASTQ-bestand van het monster. Elk item in een fastq-bestanden bestaat uit 4 regels:
- een sequentieidentificatie met informatie over de sequencing run en het cluster. De exacte inhoud van deze lijn varieert op basis van de BCL naar FASTQ conversie software gebruikt.
- de reeks (de basisaanroepen; A, C, T, G en N).
- een scheidingsteken, dat gewoon een plus (+) teken is.
- de kwaliteitsscores voor basisoproepen. Deze zijn Phred + 33 gecodeerd, met behulp van ASCII-tekens om de numerieke kwaliteitsscores weer te geven.
hier is een voorbeeld van een enkele regel in een R1 FASTQ-bestand:
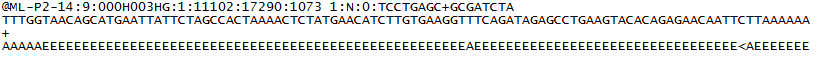
meer gedetailleerde informatie over het fastq-formaat vindt u hier.
hoe een FASTQ-bestand te bekijken
FASTQ-bestanden kunnen tot miljoenen items bevatten en kunnen meerdere megabytes of gigabytes groot zijn, waardoor ze vaak te groot zijn om in een normale teksteditor te openen. Over het algemeen is het niet nodig om FASTQ-bestanden te bekijken, omdat het intermediaire uitvoerbestanden zijn die worden gebruikt als input voor tools die downstream-analyse uitvoeren, zoals uitlijning naar een referentie of de novo-assemblage.
Als u een FASTQ-bestand wilt bekijken om problemen op te lossen of uit nieuwsgierigheid, hebt u een teksteditor nodig die zeer grote bestanden kan verwerken, of toegang tot een Unix-of Linux-systeem waar grote bestanden kunnen worden bekeken via de opdrachtregel.
hoe fastq-bestanden te genereren
FASTQ-bestanden genereren is de eerste stap voor alle analyseworkflows die worden gebruikt door MiSeq Reporter op de MiSeq en Local Run Manager op de MiniSeq. Wanneer de analyse is voltooid, bevinden de fastq-bestanden zich in <run folder>\Data\Intensities\BaseCalls op de MiSeq en <output folder>\Alignment_#\<submap>\Fastq op de MiniSeq.
voor alle runs die zijn geüpload naar de BaseSpace Sequence Hub, wordt FASTQ-bestanden automatisch gegenereerd nadat de run volledig is geüpload, en worden de fastq-bestanden gebruikt als invoer voor de verschillende analyse-apps op de BaseSpace Sequence Hub. Op BaseSpace Sequence Hub, kunt u uw FASTQ bestanden te vinden in het project(en) geassocieerd met uw run.
de bcl2fastq-conversiesoftware kan worden gebruikt om FASTQ-bestanden te genereren uit gegevens die zijn gegenereerd op alle huidige Illumina-sequentiesystemen.
voor informatie over de verschillende instellingen die kunnen worden toegepast tijdens het aanmaken van FASTQ-bestanden, zie de software gebruikershandleidingen hieronder.
- MiSeq Reporter
Local Run Manager
bcl2fastq
aanvullende informatie
- een beschrijving en vereisten voor clusters om filter te halen zijn te vinden in paragraaf 1.5.8 van de MiSeq: Imaging and Base Calling online training course.
- Zie 2-kanaals SBS-technologie voor meer informatie over base-oproepen op NovaSeq -, NextSeq 500/550-en MiniSeq-systemen.
- zie Illumina Sequencing Technology voor meer informatie over base calling op MiSeq en HiSeq systemen.