tehnologia de secvențiere Illumina utilizează chimia generării clusterelor și secvențierii prin sinteză (SBS) pentru a secvența milioane sau miliarde de clustere pe o celulă de flux, în funcție de platforma de secvențiere. În timpul chimiei SBS, pentru fiecare cluster, apelurile de bază sunt efectuate și stocate pentru fiecare ciclu de secvențiere prin software-ul de analiză în timp real (RTA) de pe instrument. RTA stochează datele apelului de bază sub formă de fișiere individuale de apel de bază (sau BCL). Când se finalizează secvențierea, apelurile de bază din fișierele BCL trebuie convertite în date de secvență. Acest proces se numește conversie BCL în FASTQ.
un fișier FASTQ este un fișier text care conține datele de secvență din clusterele care trec filtrul pe o celulă de flux (pentru mai multe informații despre clusterele care trec filtrul, consultați secțiunea „Informații suplimentare” din acest buletin). Dacă probele au fost multiplexate, primul pas în generarea de fișiere FASTQ este demultiplexarea. Demultiplexarea atribuie clustere unui eșantion, pe baza secvenței(secvențelor) indexului clusterului. După demultiplexare, secvențele asamblate sunt scrise în fișiere FASTQ pe eșantion. Dacă eșantioanele nu au fost multiplexate, etapa de demultiplexare nu are loc și, pentru fiecare bandă de celule de flux, toate clusterele sunt atribuite unui singur eșantion.
pentru o rulare cu o singură citire, se creează un fișier Fastq citit 1 (R1) pentru fiecare eșantion pe banda celulei de flux. Pentru o rulare pereche-end, un fișier R1 și o citire 2 (R2) FASTQ este creat pentru fiecare eșantion pentru fiecare bandă. Fișierele FASTQ sunt comprimate și create cu extensia*.rapid.gz.
cum arată un fișier FASTQ?
pentru fiecare cluster care trece filtrul, o singură secvență este scrisă în fișierul R1 FASTQ al eșantionului corespunzător și, pentru o rulare pereche, o singură secvență este scrisă și în fișierul R2 FASTQ al eșantionului. Fiecare intrare într-un fișiere FASTQ este format din 4 linii:
- un identificator de secvență cu informații despre rularea secvențierii și cluster. Conținutul exact al acestei linii variază în funcție de software-ul de conversie BCL la FASTQ utilizat.
- secvența (apelurile de bază; A, C, T, G și N).
- un separator, care este pur și simplu un semn plus ( + ).
- scorurile de calitate apel de bază. Acestea sunt phred +33 codificate, folosind caractere ASCII pentru a reprezenta scorurile numerice de calitate.
Iată un exemplu de intrare unică într-un fișier R1 FASTQ:
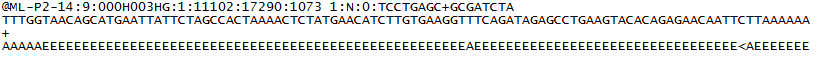
informații mai detaliate despre formatul FASTQ pot fi găsite aici.
cum se vizualizează un fișier FASTQ
fișierele FASTQ pot conține până la milioane de intrări și pot avea dimensiuni de câțiva megaocteți sau gigaocteți, ceea ce le face adesea prea mari pentru a fi deschise într-un editor de text normal. În general, nu este necesar să vizualizați fișiere FASTQ, deoarece acestea sunt fișiere de ieșire intermediare utilizate ca intrare pentru instrumente care efectuează analize în aval, cum ar fi alinierea la o referință sau un ansamblu de novo.
dacă trebuie să vizualizați un fișier FASTQ în scopuri de depanare sau din curiozitate, veți avea nevoie fie de un editor de text care să poată gestiona fișiere foarte mari, fie de acces la un sistem UNIX sau Linux unde fișierele mari pot fi vizualizate prin linia de comandă.
cum se generează fișiere FASTQ
generarea de fișiere FASTQ este primul pas pentru toate fluxurile de lucru de analiză utilizate de reporterul MiSeq pe MiSeq și local Run Manager pe MiniSeq. Când se finalizează analiza, fișierele FASTQ se află în<run folder >\Data\Intensities\BaseCalls pe MiSeq și<output folder>\Alignment_#\<subfolder >\Fastq pe MiniSeq.
pentru toate rulările încărcate în BaseSpace Sequence Hub, generarea de fișiere FASTQ are loc automat după încărcarea completă a rulării, iar fișierele FASTQ sunt utilizate ca intrare pentru diferitele aplicații de analiză de pe BaseSpace Sequence Hub. Pe BaseSpace Sequence Hub, puteți găsi fișierele FASTQ în proiectul(proiectele) asociat (e) cu rularea.
software-ul de conversie bcl2fastq poate fi utilizat pentru a genera fișiere FASTQ din datele generate pe toate sistemele actuale de secvențiere Illumina.
pentru informații despre diferitele setări care pot fi aplicate în timpul generării fișierelor FASTQ, consultați ghidurile de utilizare a software-ului de mai jos.
- MiSeq Reporter
local Run Manager
bcl2fastq
informații suplimentare
- o descriere și cerințe pentru clustere pentru a trece filtru pot fi găsite în secțiunea 1.5.8 din MiSeq: Imaging și Bază de asteptare curs de formare online.
- consultați tehnologia SBS cu 2 canale pentru mai multe informații despre apelarea bazei pe sistemele NovaSeq, NextSeq 500/550 și MiniSeq.
- consultați tehnologia de secvențiere Illumina pentru mai multe informații despre apelarea bazei pe sistemele MiSeq și HiSeq.